
So meistern Sie Ihr Product Lifecycle Reporting unter der MDR - Anwendungstipps für den gesamten Produktlebenszyklus
18.06.2024
Sie haben Fragen zum Beitrag oder möchten mehr über unsere Leistungen erfahren? Wir freuen uns auf Ihre Nachricht!Jetzt unverbindlich anfragen
Mit der Medical Device Regulation (Verordnung (EU) 2017/745 (MDR)) und den begleitenden Leitlinien der Medical Device Coordination Group (MDCG) ergeben sich entlang des gesamten Produktlebenszyklus viele Verpflichtungen für Sie als Hersteller von Medizinprodukten. Dazu gehören das regelmäßige Anfertigen von Berichten und ggf. das Aktualisieren von Plänen für die Überwachung und klinische Nachbeobachtung nach dem Inverkehrbringen, sowie für das Risikomanagement und die klinische Bewertung. Manche Aktualisierungszeitpunkte sind dabei fest vorgeschrieben, andere müssen in diesen Zyklus sinnvoll eingegliedert werden. Um allen Anforderungen im Rahmen dieses "Product Lifecycle Reporting" gerecht zu werden und alle Dokumente rechtzeitig bereitstellen zu können, müssen die einzelnen Phasen gut aufeinander abgestimmt sein.
Das Risikomanagement begleitet den gesamten Produktlebenszyklus von der Produktidee bis zum Lebensende des letzten Stücks auf dem Markt. Es muss alle Risiken identifizieren, Akzeptanzkriterien aufstellen, und das vom Produkt nach bestmöglicher Minderung ausgehende Restrisiko bewerten. Dazu benötigt es Validierungsdaten wie sie z. B. die klinische Bewertung ermittelt.Das Post-Market Surveillance (PMS)-System begleitet den Produktlebenszyklus ab Markteintritt. Es muss u. a. fortwährend gewährleisten, dass neue, geänderte oder aufkommende Risiken jederzeit erkannt und – falls nötig – angepasste und der Dringlichkeit entsprechende Maßnahmen sofort eingeleitet werden. Letztere können Produktänderungen und Feldmaßnahmen umfassen.
Bereits ab dem Zeitpunkt der Produktidee und während der Produktentwicklung muss die Gewinnung klinischer Daten geplant werden . Diese müssen dann erzeugt und analysiert werden. Nach dem klinischen Bewertungsbericht wird der PMCF-Plan (Post-Market Clinical Follow-up-Plan) erstellt. Für die CE-Kennzeichnung muss außerdem noch der PMS-Plan auf die Beine gestellt werden, und Klasse III-Produkte und Implantate benötigen überdies noch einen ersten SSCP (Summary of Safety and Clinical Performance).Für Legacy Devices gilt es zu prüfen, ob alle Dokumente vorliegen, bevor sie die CE-Kennzeichnung nach MDR erhalten können. Nicht alle Berichte waren unter der Medical Device Directive (MDD – Richtlinie 93/42/EWG über Medizinprodukte) und auch während der Übergangsphase erforderlich, weshalb sie meistens auch nicht vorliegen. Das betrifft vor allem den SSCP- und häufig ist die klinische Bewertung zu minimalistisch. Diese Berichte müssen für die Zertifizierung unter der MDR nun aber konform vorliegen. Die Datenlage für Legacy Devices muss unter den Gesichtspunkten der MDR und gemäß MDCG 2020-6 der Medical Device Coordination Group ausreichend sein.Die Forderungen für ein PMS-System inkl. Vigilanz und seine Dokumentation gemäß MDR galten auch für Legacy Devices bereits seit Inkrafttreten im Jahr 2021. Diese Pläne und Berichte sollten für die Legacy Devices also mittlerweile vorliegen.
Nach erfolgreicher CE-Kennzeichnung und Markteintritt befinden Sie sich in einem Überwachungszeitraum von 1 Jahr (Medizinprodukte der Klassen IIb und III), 2 Jahren (Klasse IIa) oder bis Bedarf für einen Bericht entsteht (Klasse I). In diesem Zeitraum werden die geplanten PMS- und PMCF-Aktivitäten durchgeführt, Daten gesammelt und Aufzeichnungen erstellt. Das PMS-System muss dabei jederzeit und sofort auf neue Informationen reagieren.Am Ende des Überwachungszeitraums werden die angefallenen Daten in entsprechend aktualisierten Berichten (PMS-Bericht/PSUR (Periodic Safety Update Report) sowie PMCF- und der anschließende klinische Bewertungsbericht) zusammengefasst und ausgewertet. Bei Bedarf werden Folgemaßnahmen im Einklang mit den PMS-Zielen definiert. Pläne müssen üblicherweise nicht aktualisiert werden, sofern sich die Planung als vollständig und geeignet erwiesen hat.Bekanntwerden neuer RisikenJederzeit können allerdings neue, geänderte oder aufkommende Risiken bekannt werden, auf die das PMS-System unter Beteiligung des Risikomanagements (und weiterer betroffener Abteilungen) reagieren muss. Hier kann es notwendig werden, den PMS-Plan zu aktualisieren, um gezielt nötige weitere Daten zu erfassen. Das neue Risiko wird der Risikoanalyse zugeführt und wenn erforderlich übernimmt auch der Clinical Evaluation Plan (CEP) das neue Risiko im Rahmen der Planung der klinischen Nutzen-Risiko Bewertung. Die zusätzlichen Maßnahmen zur Beschaffung weiterer nötiger klinischer Daten werden im PMCF-Plan definiert.Dies kann sofort geschehen, die laufenden Aktivitäten werden dann um die neuen Aspekte erweitert. Am Ende des Überwachungszeitraums werden die entsprechenden Berichte von PMS, Risikomanagement und klinischer Bewertung umfänglich aktualisiert.Wenn Änderungen an Produkten mit bestehendem MDR-Zertifikat oder an deren Zweckbestimmung eingeführt werden sollen, so wird diese Änderung wie eine Neuentwicklung betrachtet. Entsprechende zusätzliche oder erweiterte Pläne, Daten und Berichte müssen erzeugt werden. Nach erfolgreicher Konformitätsbewertung kann die Variante (oder das Nachfolgemodell) in Verkehr gebracht werden. Für das bisherige Produkt werden die bestehenden Berichte weiterhin bis zu dessen Lebensende regelmäßig aktualisiert. Eine Zusammenlegung in den Dokumenten ist natürlich möglich.
Die Anforderungen an das PMS-System gelten für die gesamte Lebensdauer aller Produkte bis zum vom Hersteller definierten Ende des letzten auf dem Markt befindlichen Stücks. Dies gilt auch für eine regelmäßige Berichterstattung. Das PMS-System muss daher bis zum letzten Moment, in dem noch z. B. mit einer Field Safety Corrective Action (FSCA / Sicherheitskorrekturmaßnahmen im Feld) reagiert werden kann, aktiv bleiben.Möglicherweise sammeln Sie in dieser Zeit genügend klinische Daten aus den PMCF-Aktivitäten für Ihr Produkt, um nach und nach sämtliche nötigen klinischen Schlussfolgerungen für die gesamte Lebensdauer gesichert und ohne weitere Annahmen treffen zu können. Hier können Sie an eine Aufdehnung der Aktualisierungszeiten für den Clinical Evaluation Report (CER) denken. Spätestens zum Verkaufsstopp des Produkts können Sie mit entsprechender Begründung Ihren letzten CER schreiben. Eine Designänderung als Maßnahme auf Probleme wird nach einem Verkaufsstopp nicht mehr stattfinden und für alle weiteren reaktiven Maßnahmen bleibt das PMS-System aktiv. Eine Wiederaufnahme des CERs fände allerdings statt, wenn sich durch eine neue Lage das Nutzen/Risiko-Verhältnis ins Inakzeptable dreht. Der PMCF-Report wird für PMS benötigt und muss daher weitergeführt werden. So bleibt auch die Reaktionsfähigkeit auf unerwartete klinische Daten bestehen.
Das folgende Schaubild stellt die erläuterten Phasen des Produktlebenszyklus, also die Entwicklung (blau) und die Zeit nach dem Inverkehrbringen (orange), sowie die erwähnten Pläne und Berichte in ihrer logischen Abfolge dar - am Beispiel eines neu entwickelten Produkts. So fällt es leichter, die Abhängigkeiten zu erkennen, auf die wir im Folgenden eingehen werden.
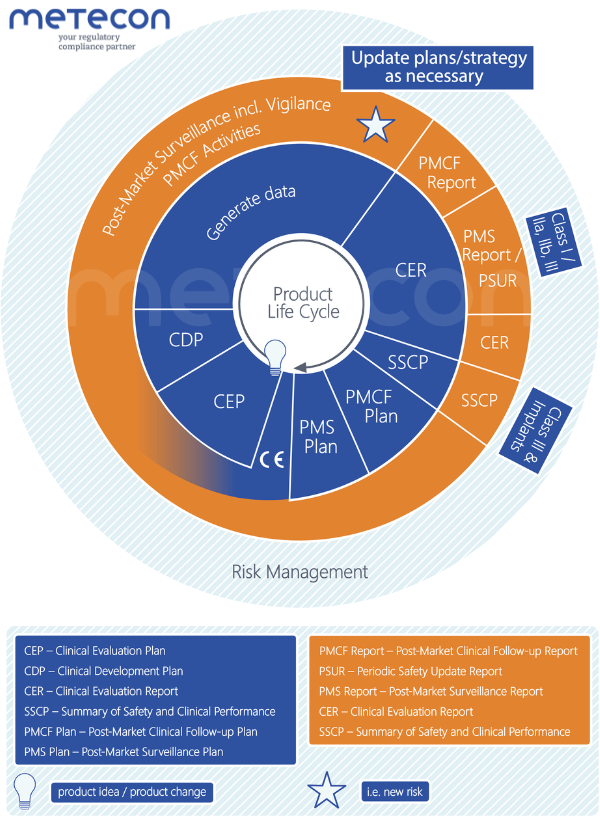
Der Clinical Evaluation Plan legt den Umfang des klinischen Nachweises anhand der zu bedienenden relevanten Grundlegenden Sicherheits- und Leistungsanforderungen (general safety and performance requirements (GSPR)) in Anhang I der MDR fest. Dabei werden die zu analysierenden Aspekte der Zweckbestimmung (und damit der intendierten klinischen Leistung und des klinischen Nutzens) und der Restrisiken gemäß Risikoanalyse festgelegt. Die benötigten Parameter zur Bewertung des Nutzen-Risiko-Verhältnisses werden identifiziert. Alle Werbeaussagen müssen ebenfalls nachgewiesen werden und benötigen gegebenenfalls weitere Parameter.Unter Berücksichtigung dieser Aspekte und Parameter, der bereits vorhandenen prä- und klinischen Daten, der Risikoklasse und möglicher Äquivalenzprodukte wird die Strategie (Route) der klinischen Bewertung festgelegt.Der CEP wird aktualisiert, wenn sich der notwendige Umfang oder die Route ändert.Schließlich wird an den Parametern und der Route die Suchstrategie ausgerichtet, nach der die benötigten Daten zum Produkt erhoben werden. Zu dieser Strategie gehört mindestens eine Literatursuche (mit Planung der Quellen, Suchbegriffe, Selektions- und Bewertungskriterien) und wo notwendig eine eigene klinische Studie. Mit einer weiteren separaten Suchstrategie wird die Erarbeitung des State of the art (SOTA) geplant, die insbesondere die Identifizierung der etablierten Anforderung an Nutzen und Leistung und Akzeptanz von Risiken zum Ziel hat.
Der klinische Entwicklungsplan (CDP) ist formal ein Teil des CEP. Er legt zu Beginn des Entwicklungsprozesses dar, wie Sie benötigte eigene klinische Daten erzeugen. Dies kann explorative Studien, Studien zur Erstanwendung am Menschen, Durchführbarkeitsstudien und Pilotstudien bis hin zu Belegstudien beinhalten; ein Ausblick auf mögliche PMCF-Aktivitäten ist an dieser Stelle ebenfalls bereits möglich.Der CDP bestimmt anhand der zu bedienenden Parameter die Ziele und Endpunkte der Studie(n). Er gibt außerdem Akzeptanzkriterien, Meilensteine, ein mögliches Studiendesign und den Zeitrahmen vor. Der CDP ist ein übersichtliches Dachdokument; die detaillierte Planung jeder Studie findet sich im jeweiligen Clinical Investigation Protocol (CIP).Der CDP wird aktualisiert, wenn Studienziele geändert oder weitere Studien hinzukommen.
Der klinische Evaluierungsbericht (CER) stellt die Ergebnisse der Bewertung aller klinischen Daten dar. Dabei werden Daten des Produkts oder nachgewiesenermaßen äquivalenter Produkte gesammelt, selektiert, bewertet und analysiert. Es wird geprüft, ob und wie sehr die Anforderungen an Leistung/Nutzen und Sicherheit (Risiken und unerwünschte Nebenwirkungen) erfüllt werden. Daraufhin wird die Vertretbarkeit des sich ergebenden Nutzen/Risiko-Verhältnisses bewertet. Für eine Schlussfolgerung zur Konformität mit den GSPRs sind ausreichende bestätigende klinische Daten notwendig. Zuletzt identifiziert der CER den Bedarf weiterer klinischer Daten, die im PMCF erhoben werden sollen.Der CER wird kontinuierlich und regelmäßig mit den PMCF-Daten aktualisiert, um die fortgesetzte Konformität mit den grundlegenden Sicherheits- und Leistungsanforderungen zu bewerten.
Das Post-Market Clinical Follow-up (PMCF) ist formal Teil der Post-Market Surveillance. Dabei soll das PMCF die bisher ausreichende klinische Evidenz zum Produkt für die vollständige Lebensdauer erweitern. PMCF beantwortet auch offene Fragen, die im Rahmen der Klinischen Bewertung bisher nur geschätzt werden konnten (z. B. Langzeitverhalten, Überwachung von Nebenwirkungen und Kontraindikationen). Mit diesen Daten wird die klinische Evaluierung kontinuierlich und regelmäßig aktualisiert.Der PMCF-Plan beschreibt gemäß MDCG 2020-7 die Methoden und Verfahren für das proaktive Sammeln oder Erzeugen klinischer Daten. Er arbeitet Aktivitäten aus, die sowohl zu ausgewählten Parametern weitere Daten suchen als auch der generellen Überwachung und Bestätigung von Anwendung, Leistung und Sicherheit dienen. Der Umfang kann je nach den dazu erforderlichen PMCF-Aktivitäten variieren. Daher kann an dieser Stelle ein PMCF-Masterplan erstellt werden, der auf verschiedene weitere Pläne verweist, in denen die Einzelaktivitäten detailliert dargestellt sind.Der PMCF-Plan wird aktualisiert, wenn Anpassungen der Aktivitäten im PMCF-Report als notwendig erkannt werden, oder wenn der CER geänderte oder zusätzliche Anforderungen an das PMCF ermittelt hat.
Im Post-Market Surveillance-Plan definiert der Hersteller, auf welche Art und Weise das Produkt nach dem Inverkehrbringen überwacht wird und welche Daten erhoben bzw. gesammelt werden sollen. Bei der produktspezifischen Planung der Aktivitäten sind die Eigenschaften und die damit verbundenen Risiken des Produkts und die Erkenntnisse aus der Klinischen Bewertung zu berücksichtigen. Des Weiteren wird festgelegt und definiert, nach welchen Methoden die Auswertung der Daten erfolgt und welche Kriterien für die Auswertung verwendet werden. Die Datenerhebung am Markt muss proaktiv durch den Hersteller erfolgen. Wichtig ist, dass die Planung die PMS-Ziele aus Artikel 83 der MDR befolgt.Der PMS-Plan muss aktualisiert werden, wenn es neue zu berücksichtigende Aspekte gibt (z. B. ein neuer versprochener klinischer Nutzen), wenn in der aktuellen Planung Verbesserungsmöglichkeiten festgestellt werden oder wenn es eine Änderung bei den vom PMS-Plan erfassten Geräten gibt. Der PMS-Plan muss auch dann aktualisiert werden, wenn neue Datensätze verfügbar sind, die in das PMS einbezogen werden sollten. Es gibt weitere Situationen, in denen formale Änderungen des PMS-Plans erforderlich sind.Proaktives PMSBei der Planung der proaktiven PMS-Aktivitäten muss man berücksichtigen, welche Aktivitäten als PMCF-Aktivitäten durchgeführt werden sollen. Dies ist wichtig, da die PMCF-Aktivitäten gemäß Anhang XIV der MDR geplant werden, den PMCF-Zielen entsprechen sollen und in einem PMCF-Bewertungsbericht zusammengefasst werden. Wenn eine Aktivität als Teil der PMCF durchgeführt wird, dann sollten die erhobenen Daten der Definition von klinischen Daten gemäß Artikel 2(48) der MDR entsprechen. Das bedeutet, dass nicht jede proaktive Tätigkeit, die im Rahmen des PMS durchgeführt wird, die Kriterien für PMCF erfüllen wird. Die Entscheidung zur Differenzierung zwischen einer proaktiven PMS-Tätigkeit und einer PMCF-Tätigkeit sollte in einem entsprechenden Prozess geregelt werden. Eine falsche Umsetzung zeigt sich, wenn die PMCF-Daten keine klinische Relevanz haben.Typische proaktive PMS- oder PMCF-Datensätze sind eine Recherche der wissenschaftlichen Fachliteratur und veröffentlichte Berichte über sonstige klinische Erfahrungen entweder mit dem eigenen oder mit gleichartigen Produkten. Die Erfahrung für gleichartige Produkte wird auch in Rahmen einer Datenbanksuche zu Vorkommnissen oder Sicherheitskorrekturmaßnahmen im Feld (FSCAs) untersucht, häufig wird hier die Manufacturer and User Facility Device Experience (MAUDE) Datenbank der US-amerikanischen Food & Drug Administration (FDA) eingesetzt. Anwenderbefragungen können entweder als proaktive PMS- oder PMCF-Aktivität durchgeführt werden, je nachdem, was der Umfrageziel ist.Es ist wichtig, dass eine Anwenderbefragung im Rahmen von PMS/PMCF ordentlich geplant und durchgeführt wird, d.h. von der Zielsetzung der Umfrage bis zu der Datenauswertung. Falls eine Anwenderbefragung mit Software unterstützt wird, sollte diese Software auch für diesen Zweck geeignet sein. Hier sind mehrere Themen zu betrachten, wie Datenfelder (Freitext oder Auswahl), Sprache, Datensicherheit und Auswertungsmöglichkeiten.Reaktives PMSIn der Regel werden reaktive Datensätze in den PMS-Plan und nicht in den PMCF-Plan aufgenommen und umfassen mehrere der in Anhang III der MDR erwähnten Datensätze. Zu diesen Datensätzen gehören beispielsweise schwerwiegende Vorkommnisse, Sicherheitskorrekturmaßnahmen im Feld und Trendberichte gemäß Artikel 88 der MDR. Im Rahmen des PMS werden Informationen und Ereignisse unmittelbar nach ihrem Eingang ausgewertet und die entsprechenden Folgemaßnahmen direkt eingeleitet. In diesem Fall sind die Daten, die dann im Bericht enthalten sind, eine Zusammenfassung der Ereignisse während des Überwachungszeitraums einschließlich aller erforderlichen Folgemaßnahmen.Im PMS-Plan sollten die benötigten Informationen nach MDCG 2022-21 geplant werden, damit die Daten vollständig in den Berichten präsentiert werden könnten. Wie bei den proaktiven Daten sollte der PMS-Plan auch hier beschreiben, wie die Datenanalyse und -bewertung durchgeführt werden soll.
Die klinischen Daten und ggf. Zwischenergebnisse aus den PMCF-Aktivitäten zum eigenen Produkt sowie zu Äquivalenz- und ähnlichen Produkten werden gemäß MDCG 2020-8 zusammengefasst und präsentiert. Die Daten werden bewertet, ob sie die bisherigen Schlussfolgerungen in der klinischen Bewertung potenziell bestätigen oder einschränken, oder ob sie als neue Informationen einen Einfluss auf die bisherige Bewertung haben. Die Daten werden auf Qualität und die Aktivitäten auf Effektivität geprüft. Der PMCF-Report kann auch über Folgemaßnahmen entscheiden.Der PMCF-Report wird regelmäßig neu erstellt. Die Aktualisierungszeiten folgen der Berichterstattung in der PMS.
Der PMS-Report gemäß Artikel 85 der MDR wird für Klasse I-Produkte erstellt und beinhaltet die Zusammenfassung der Ergebnisse aus den gewonnenen PMS-Daten (inklusive der PMCF-Daten) des Überwachungszeitraums. Auf Basis dieser Ergebnisse wird eine Schlussfolgerung getroffen, dieu. a. an die Klinische Bewertung weitergegeben wird. Zudem werden Korrektur- und Vorbeugemaßnahmen dargelegt und begründet. Sinn und Zweck des PMS-Reports ist es, über die gesamte Produktlebensdauer Erkenntnisse über das Verhalten des Produkts am Markt zu gewinnen, die für die weitere Produktentwicklung sowie die Gewährleistung der Produktsicherheit genutzt werden können. Auf diese Weise kann sichergestellt werden, dass das Produkt zu jeder Zeit den Anforderungen der Verordnung entspricht.Der Periodic Safety Update Report (PSUR) wird für Klasse IIa-, IIb- und III-Produkte erstellt und beinhaltet ebenso wie der PMS-Bericht die Zusammenfassung der Ergebnisse aus den gewonnen Marktdaten des letzten Überwachungszeitraums. Auch hier wird auf Basis dieser Ergebnisse eine Schlussfolgerung getroffen, die u. a. an die Klinische Bewertung weitergegeben wird. Zudem werden Korrektur- und Vorbeugemaßnahmen (Corrective and Preventive Action (CAPA)) dargelegt und begründet. Neben den Themen des PMS-Berichts beinhaltet der PSUR zusätzlich die Schlussfolgerungen aus der Nutzen-Risiko-Abwägung, die wichtigsten Ergebnisse aus dem PMCF und die Gesamtabsatzmenge des Produkts sowie weitere Angaben, z. B. zu Häufigkeiten der Anwendung.Sinn und Zweck des PSURs ist es, wie in der MDCG 2022-21 aufgeführt, dass der PSUR einen allgemeinen Überblick geben und alle Ergebnisse und Schlussfolgerungen zusammenfassen sollte. Ganz wichtig bei der PSUR-Erstellung ist eine Aussage bezüglich der Datenvalidität zu treffen, um mit Einschränkungen transparent umzugehen. Schlussfolgerungen werden auch getroffen über z.B. neue Risiken oder Nutzen und mögliche negative Auswirkungen auf die Nutzen-Risiko-Abwägung (siehe Anhang I der MDCG 2022-21). Die PMCF-Schlussfolgerungen sind dabei ein wesentlicher Beitrag zur fortgesetzten Feststellung eines akzeptablen Nutzen-Risiko-Profils, und betont die Wichtigkeit, PMCF richtig ins PMS zu integrieren.Der PSUR wird regelmäßig neu erstellt (auch für Legacy Devices). Die Aktualisierungszeiten folgen der Risikoklasse. Der PMS-Bericht (für Klasse I Produkte) wird bei Bedarf erstellt und sollte sich an der Aktualisierung der klinischen Bewertung orientieren.
Der SSCP-Bericht muss für Klasse III- und implantierbare Produkte gemäß Artikel 32 der MDR und MDCG 2019-9 Rev.1 angefertigt werden. Sinn dieses Kurzberichts ist es, das Produkt im Kontext seiner Anwendung darzustellen, Restrisiken und unerwünschte Wirkungen auszuweisen sowie alternative Optionen in Bezug auf Therapie oder Diagnose aufzuzeigen.Der Bericht wird in der Gebrauchsanweisung oder der Kennzeichnung ausgewiesen und wird der Öffentlichkeit über EUDAMED zugänglich gemacht. Dabei muss der Bericht so geschrieben sein, dass er auch für den Laien verständlich ist, sofern diese die Anwender sind.Vor Veröffentlichung erfolgt eine Validierung des Berichts durch die Benannte Stelle, die den Bericht dann in EUDAMED bereitstellt.Der SSCP wird jährlich aktualisiert.
Im Überwachungszeitraum finden alle geplanten Aktivitäten zur Erfassung der Daten statt, anhand derer die fortgesetzte Leistungsfähigkeit und Sicherheit bewertet werden können. Gegen Ende dieses Zeitraums beginnt die zugehörige Berichterstattung. Diese Phase sollte kompakt gehalten werden, so dass sich keiner der Berichte im Übermaß verspätet. Während der Schreibphase bleibt das PMS-System selbstverständlich aktiv, und Pläne werden grundsätzlich geändert, sobald es nötig wird. Zusätzliche Aktivitäten werden den laufenden sofort hinzugefügt.Einen reibungslosen Ablauf, der die gegenseitige Bezugnahme der dabei erstellten Dokumente berücksichtigt, haben wir Ihnen hier vorgestellt. Im Umfang dieses Blogs können wir aber nicht jede individuelle Begebenheit berücksichtigen. Außerdem können jederzeit spezielle Situationen auftreten, wiez. B. oben erwähnte neue Risiken, aber auch eine neue Normenlage, oder ein Zeitpunkt, an dem Sie Ihre PMCF-Aktivitäten verbessern müssen (oder beenden können).Arbeiten Sie trotzdem zunächst darauf hin, den dargestellten synchronisierten Rhythmus zu erreichen und dann Ihre Berichte immer zeitnah zu pflegen. Vielleicht benötigen Sie dazu auch eine zusätzliche Iteration.
Unsere Erfahrung zeigt, dass es zahlreiche Sondersituationen geben kann, die den angestrebten synchronisierten Rhythmus aller Berichte erschweren. Bei Ihnen ist dies der Fall? Unser Team erarbeitet mit Ihnen eine Strategie für Ihr Product Lifecycle Reporting, die Ihrem Produkt und Ihren Anforderungen entspricht und die ihnen hilft, auch auf Unvorhergesehenes souverän zu reagieren. Schreiben Sie uns eine Nachricht und finden Sie schnell und unkompliziert heraus, wie wir Ihnen weiterhelfen können!
 Dr. Jennifer Dean
Dr. Jennifer Dean
Phasen des Produktlebenszyklus unter der MDR
Das Risikomanagement begleitet den gesamten Produktlebenszyklus von der Produktidee bis zum Lebensende des letzten Stücks auf dem Markt. Es muss alle Risiken identifizieren, Akzeptanzkriterien aufstellen, und das vom Produkt nach bestmöglicher Minderung ausgehende Restrisiko bewerten. Dazu benötigt es Validierungsdaten wie sie z. B. die klinische Bewertung ermittelt.Das Post-Market Surveillance (PMS)-System begleitet den Produktlebenszyklus ab Markteintritt. Es muss u. a. fortwährend gewährleisten, dass neue, geänderte oder aufkommende Risiken jederzeit erkannt und – falls nötig – angepasste und der Dringlichkeit entsprechende Maßnahmen sofort eingeleitet werden. Letztere können Produktänderungen und Feldmaßnahmen umfassen.
Entwicklung und Legacy Devices
Bereits ab dem Zeitpunkt der Produktidee und während der Produktentwicklung muss die Gewinnung klinischer Daten geplant werden . Diese müssen dann erzeugt und analysiert werden. Nach dem klinischen Bewertungsbericht wird der PMCF-Plan (Post-Market Clinical Follow-up-Plan) erstellt. Für die CE-Kennzeichnung muss außerdem noch der PMS-Plan auf die Beine gestellt werden, und Klasse III-Produkte und Implantate benötigen überdies noch einen ersten SSCP (Summary of Safety and Clinical Performance).Für Legacy Devices gilt es zu prüfen, ob alle Dokumente vorliegen, bevor sie die CE-Kennzeichnung nach MDR erhalten können. Nicht alle Berichte waren unter der Medical Device Directive (MDD – Richtlinie 93/42/EWG über Medizinprodukte) und auch während der Übergangsphase erforderlich, weshalb sie meistens auch nicht vorliegen. Das betrifft vor allem den SSCP- und häufig ist die klinische Bewertung zu minimalistisch. Diese Berichte müssen für die Zertifizierung unter der MDR nun aber konform vorliegen. Die Datenlage für Legacy Devices muss unter den Gesichtspunkten der MDR und gemäß MDCG 2020-6 der Medical Device Coordination Group ausreichend sein.Die Forderungen für ein PMS-System inkl. Vigilanz und seine Dokumentation gemäß MDR galten auch für Legacy Devices bereits seit Inkrafttreten im Jahr 2021. Diese Pläne und Berichte sollten für die Legacy Devices also mittlerweile vorliegen.
Nach dem Inverkehrbringen unter der MDR
Nach erfolgreicher CE-Kennzeichnung und Markteintritt befinden Sie sich in einem Überwachungszeitraum von 1 Jahr (Medizinprodukte der Klassen IIb und III), 2 Jahren (Klasse IIa) oder bis Bedarf für einen Bericht entsteht (Klasse I). In diesem Zeitraum werden die geplanten PMS- und PMCF-Aktivitäten durchgeführt, Daten gesammelt und Aufzeichnungen erstellt. Das PMS-System muss dabei jederzeit und sofort auf neue Informationen reagieren.Am Ende des Überwachungszeitraums werden die angefallenen Daten in entsprechend aktualisierten Berichten (PMS-Bericht/PSUR (Periodic Safety Update Report) sowie PMCF- und der anschließende klinische Bewertungsbericht) zusammengefasst und ausgewertet. Bei Bedarf werden Folgemaßnahmen im Einklang mit den PMS-Zielen definiert. Pläne müssen üblicherweise nicht aktualisiert werden, sofern sich die Planung als vollständig und geeignet erwiesen hat.Bekanntwerden neuer RisikenJederzeit können allerdings neue, geänderte oder aufkommende Risiken bekannt werden, auf die das PMS-System unter Beteiligung des Risikomanagements (und weiterer betroffener Abteilungen) reagieren muss. Hier kann es notwendig werden, den PMS-Plan zu aktualisieren, um gezielt nötige weitere Daten zu erfassen. Das neue Risiko wird der Risikoanalyse zugeführt und wenn erforderlich übernimmt auch der Clinical Evaluation Plan (CEP) das neue Risiko im Rahmen der Planung der klinischen Nutzen-Risiko Bewertung. Die zusätzlichen Maßnahmen zur Beschaffung weiterer nötiger klinischer Daten werden im PMCF-Plan definiert.Dies kann sofort geschehen, die laufenden Aktivitäten werden dann um die neuen Aspekte erweitert. Am Ende des Überwachungszeitraums werden die entsprechenden Berichte von PMS, Risikomanagement und klinischer Bewertung umfänglich aktualisiert.Wenn Änderungen an Produkten mit bestehendem MDR-Zertifikat oder an deren Zweckbestimmung eingeführt werden sollen, so wird diese Änderung wie eine Neuentwicklung betrachtet. Entsprechende zusätzliche oder erweiterte Pläne, Daten und Berichte müssen erzeugt werden. Nach erfolgreicher Konformitätsbewertung kann die Variante (oder das Nachfolgemodell) in Verkehr gebracht werden. Für das bisherige Produkt werden die bestehenden Berichte weiterhin bis zu dessen Lebensende regelmäßig aktualisiert. Eine Zusammenlegung in den Dokumenten ist natürlich möglich.
Lebensende
Die Anforderungen an das PMS-System gelten für die gesamte Lebensdauer aller Produkte bis zum vom Hersteller definierten Ende des letzten auf dem Markt befindlichen Stücks. Dies gilt auch für eine regelmäßige Berichterstattung. Das PMS-System muss daher bis zum letzten Moment, in dem noch z. B. mit einer Field Safety Corrective Action (FSCA / Sicherheitskorrekturmaßnahmen im Feld) reagiert werden kann, aktiv bleiben.Möglicherweise sammeln Sie in dieser Zeit genügend klinische Daten aus den PMCF-Aktivitäten für Ihr Produkt, um nach und nach sämtliche nötigen klinischen Schlussfolgerungen für die gesamte Lebensdauer gesichert und ohne weitere Annahmen treffen zu können. Hier können Sie an eine Aufdehnung der Aktualisierungszeiten für den Clinical Evaluation Report (CER) denken. Spätestens zum Verkaufsstopp des Produkts können Sie mit entsprechender Begründung Ihren letzten CER schreiben. Eine Designänderung als Maßnahme auf Probleme wird nach einem Verkaufsstopp nicht mehr stattfinden und für alle weiteren reaktiven Maßnahmen bleibt das PMS-System aktiv. Eine Wiederaufnahme des CERs fände allerdings statt, wenn sich durch eine neue Lage das Nutzen/Risiko-Verhältnis ins Inakzeptable dreht. Der PMCF-Report wird für PMS benötigt und muss daher weitergeführt werden. So bleibt auch die Reaktionsfähigkeit auf unerwartete klinische Daten bestehen.
Pläne und Berichte
Das folgende Schaubild stellt die erläuterten Phasen des Produktlebenszyklus, also die Entwicklung (blau) und die Zeit nach dem Inverkehrbringen (orange), sowie die erwähnten Pläne und Berichte in ihrer logischen Abfolge dar - am Beispiel eines neu entwickelten Produkts. So fällt es leichter, die Abhängigkeiten zu erkennen, auf die wir im Folgenden eingehen werden.
Clinical Evaluation Plan (CEP)
Der Clinical Evaluation Plan legt den Umfang des klinischen Nachweises anhand der zu bedienenden relevanten Grundlegenden Sicherheits- und Leistungsanforderungen (general safety and performance requirements (GSPR)) in Anhang I der MDR fest. Dabei werden die zu analysierenden Aspekte der Zweckbestimmung (und damit der intendierten klinischen Leistung und des klinischen Nutzens) und der Restrisiken gemäß Risikoanalyse festgelegt. Die benötigten Parameter zur Bewertung des Nutzen-Risiko-Verhältnisses werden identifiziert. Alle Werbeaussagen müssen ebenfalls nachgewiesen werden und benötigen gegebenenfalls weitere Parameter.Unter Berücksichtigung dieser Aspekte und Parameter, der bereits vorhandenen prä- und klinischen Daten, der Risikoklasse und möglicher Äquivalenzprodukte wird die Strategie (Route) der klinischen Bewertung festgelegt.Der CEP wird aktualisiert, wenn sich der notwendige Umfang oder die Route ändert.Schließlich wird an den Parametern und der Route die Suchstrategie ausgerichtet, nach der die benötigten Daten zum Produkt erhoben werden. Zu dieser Strategie gehört mindestens eine Literatursuche (mit Planung der Quellen, Suchbegriffe, Selektions- und Bewertungskriterien) und wo notwendig eine eigene klinische Studie. Mit einer weiteren separaten Suchstrategie wird die Erarbeitung des State of the art (SOTA) geplant, die insbesondere die Identifizierung der etablierten Anforderung an Nutzen und Leistung und Akzeptanz von Risiken zum Ziel hat.
Clinical Development Plan (CDP)
Der klinische Entwicklungsplan (CDP) ist formal ein Teil des CEP. Er legt zu Beginn des Entwicklungsprozesses dar, wie Sie benötigte eigene klinische Daten erzeugen. Dies kann explorative Studien, Studien zur Erstanwendung am Menschen, Durchführbarkeitsstudien und Pilotstudien bis hin zu Belegstudien beinhalten; ein Ausblick auf mögliche PMCF-Aktivitäten ist an dieser Stelle ebenfalls bereits möglich.Der CDP bestimmt anhand der zu bedienenden Parameter die Ziele und Endpunkte der Studie(n). Er gibt außerdem Akzeptanzkriterien, Meilensteine, ein mögliches Studiendesign und den Zeitrahmen vor. Der CDP ist ein übersichtliches Dachdokument; die detaillierte Planung jeder Studie findet sich im jeweiligen Clinical Investigation Protocol (CIP).Der CDP wird aktualisiert, wenn Studienziele geändert oder weitere Studien hinzukommen.
Clinical Evaluation Report (CER)
Der klinische Evaluierungsbericht (CER) stellt die Ergebnisse der Bewertung aller klinischen Daten dar. Dabei werden Daten des Produkts oder nachgewiesenermaßen äquivalenter Produkte gesammelt, selektiert, bewertet und analysiert. Es wird geprüft, ob und wie sehr die Anforderungen an Leistung/Nutzen und Sicherheit (Risiken und unerwünschte Nebenwirkungen) erfüllt werden. Daraufhin wird die Vertretbarkeit des sich ergebenden Nutzen/Risiko-Verhältnisses bewertet. Für eine Schlussfolgerung zur Konformität mit den GSPRs sind ausreichende bestätigende klinische Daten notwendig. Zuletzt identifiziert der CER den Bedarf weiterer klinischer Daten, die im PMCF erhoben werden sollen.Der CER wird kontinuierlich und regelmäßig mit den PMCF-Daten aktualisiert, um die fortgesetzte Konformität mit den grundlegenden Sicherheits- und Leistungsanforderungen zu bewerten.
PMCF-Plan
Das Post-Market Clinical Follow-up (PMCF) ist formal Teil der Post-Market Surveillance. Dabei soll das PMCF die bisher ausreichende klinische Evidenz zum Produkt für die vollständige Lebensdauer erweitern. PMCF beantwortet auch offene Fragen, die im Rahmen der Klinischen Bewertung bisher nur geschätzt werden konnten (z. B. Langzeitverhalten, Überwachung von Nebenwirkungen und Kontraindikationen). Mit diesen Daten wird die klinische Evaluierung kontinuierlich und regelmäßig aktualisiert.Der PMCF-Plan beschreibt gemäß MDCG 2020-7 die Methoden und Verfahren für das proaktive Sammeln oder Erzeugen klinischer Daten. Er arbeitet Aktivitäten aus, die sowohl zu ausgewählten Parametern weitere Daten suchen als auch der generellen Überwachung und Bestätigung von Anwendung, Leistung und Sicherheit dienen. Der Umfang kann je nach den dazu erforderlichen PMCF-Aktivitäten variieren. Daher kann an dieser Stelle ein PMCF-Masterplan erstellt werden, der auf verschiedene weitere Pläne verweist, in denen die Einzelaktivitäten detailliert dargestellt sind.Der PMCF-Plan wird aktualisiert, wenn Anpassungen der Aktivitäten im PMCF-Report als notwendig erkannt werden, oder wenn der CER geänderte oder zusätzliche Anforderungen an das PMCF ermittelt hat.
PMS-Plan
Im Post-Market Surveillance-Plan definiert der Hersteller, auf welche Art und Weise das Produkt nach dem Inverkehrbringen überwacht wird und welche Daten erhoben bzw. gesammelt werden sollen. Bei der produktspezifischen Planung der Aktivitäten sind die Eigenschaften und die damit verbundenen Risiken des Produkts und die Erkenntnisse aus der Klinischen Bewertung zu berücksichtigen. Des Weiteren wird festgelegt und definiert, nach welchen Methoden die Auswertung der Daten erfolgt und welche Kriterien für die Auswertung verwendet werden. Die Datenerhebung am Markt muss proaktiv durch den Hersteller erfolgen. Wichtig ist, dass die Planung die PMS-Ziele aus Artikel 83 der MDR befolgt.Der PMS-Plan muss aktualisiert werden, wenn es neue zu berücksichtigende Aspekte gibt (z. B. ein neuer versprochener klinischer Nutzen), wenn in der aktuellen Planung Verbesserungsmöglichkeiten festgestellt werden oder wenn es eine Änderung bei den vom PMS-Plan erfassten Geräten gibt. Der PMS-Plan muss auch dann aktualisiert werden, wenn neue Datensätze verfügbar sind, die in das PMS einbezogen werden sollten. Es gibt weitere Situationen, in denen formale Änderungen des PMS-Plans erforderlich sind.Proaktives PMSBei der Planung der proaktiven PMS-Aktivitäten muss man berücksichtigen, welche Aktivitäten als PMCF-Aktivitäten durchgeführt werden sollen. Dies ist wichtig, da die PMCF-Aktivitäten gemäß Anhang XIV der MDR geplant werden, den PMCF-Zielen entsprechen sollen und in einem PMCF-Bewertungsbericht zusammengefasst werden. Wenn eine Aktivität als Teil der PMCF durchgeführt wird, dann sollten die erhobenen Daten der Definition von klinischen Daten gemäß Artikel 2(48) der MDR entsprechen. Das bedeutet, dass nicht jede proaktive Tätigkeit, die im Rahmen des PMS durchgeführt wird, die Kriterien für PMCF erfüllen wird. Die Entscheidung zur Differenzierung zwischen einer proaktiven PMS-Tätigkeit und einer PMCF-Tätigkeit sollte in einem entsprechenden Prozess geregelt werden. Eine falsche Umsetzung zeigt sich, wenn die PMCF-Daten keine klinische Relevanz haben.Typische proaktive PMS- oder PMCF-Datensätze sind eine Recherche der wissenschaftlichen Fachliteratur und veröffentlichte Berichte über sonstige klinische Erfahrungen entweder mit dem eigenen oder mit gleichartigen Produkten. Die Erfahrung für gleichartige Produkte wird auch in Rahmen einer Datenbanksuche zu Vorkommnissen oder Sicherheitskorrekturmaßnahmen im Feld (FSCAs) untersucht, häufig wird hier die Manufacturer and User Facility Device Experience (MAUDE) Datenbank der US-amerikanischen Food & Drug Administration (FDA) eingesetzt. Anwenderbefragungen können entweder als proaktive PMS- oder PMCF-Aktivität durchgeführt werden, je nachdem, was der Umfrageziel ist.Es ist wichtig, dass eine Anwenderbefragung im Rahmen von PMS/PMCF ordentlich geplant und durchgeführt wird, d.h. von der Zielsetzung der Umfrage bis zu der Datenauswertung. Falls eine Anwenderbefragung mit Software unterstützt wird, sollte diese Software auch für diesen Zweck geeignet sein. Hier sind mehrere Themen zu betrachten, wie Datenfelder (Freitext oder Auswahl), Sprache, Datensicherheit und Auswertungsmöglichkeiten.Reaktives PMSIn der Regel werden reaktive Datensätze in den PMS-Plan und nicht in den PMCF-Plan aufgenommen und umfassen mehrere der in Anhang III der MDR erwähnten Datensätze. Zu diesen Datensätzen gehören beispielsweise schwerwiegende Vorkommnisse, Sicherheitskorrekturmaßnahmen im Feld und Trendberichte gemäß Artikel 88 der MDR. Im Rahmen des PMS werden Informationen und Ereignisse unmittelbar nach ihrem Eingang ausgewertet und die entsprechenden Folgemaßnahmen direkt eingeleitet. In diesem Fall sind die Daten, die dann im Bericht enthalten sind, eine Zusammenfassung der Ereignisse während des Überwachungszeitraums einschließlich aller erforderlichen Folgemaßnahmen.Im PMS-Plan sollten die benötigten Informationen nach MDCG 2022-21 geplant werden, damit die Daten vollständig in den Berichten präsentiert werden könnten. Wie bei den proaktiven Daten sollte der PMS-Plan auch hier beschreiben, wie die Datenanalyse und -bewertung durchgeführt werden soll.
PMCF-Report
Die klinischen Daten und ggf. Zwischenergebnisse aus den PMCF-Aktivitäten zum eigenen Produkt sowie zu Äquivalenz- und ähnlichen Produkten werden gemäß MDCG 2020-8 zusammengefasst und präsentiert. Die Daten werden bewertet, ob sie die bisherigen Schlussfolgerungen in der klinischen Bewertung potenziell bestätigen oder einschränken, oder ob sie als neue Informationen einen Einfluss auf die bisherige Bewertung haben. Die Daten werden auf Qualität und die Aktivitäten auf Effektivität geprüft. Der PMCF-Report kann auch über Folgemaßnahmen entscheiden.Der PMCF-Report wird regelmäßig neu erstellt. Die Aktualisierungszeiten folgen der Berichterstattung in der PMS.
PMS-Report / PSUR
Der PMS-Report gemäß Artikel 85 der MDR wird für Klasse I-Produkte erstellt und beinhaltet die Zusammenfassung der Ergebnisse aus den gewonnenen PMS-Daten (inklusive der PMCF-Daten) des Überwachungszeitraums. Auf Basis dieser Ergebnisse wird eine Schlussfolgerung getroffen, die
Summary of Safety and Clinical Performance (SSCP)
Der SSCP-Bericht muss für Klasse III- und implantierbare Produkte gemäß Artikel 32 der MDR und MDCG 2019-9 Rev.1 angefertigt werden. Sinn dieses Kurzberichts ist es, das Produkt im Kontext seiner Anwendung darzustellen, Restrisiken und unerwünschte Wirkungen auszuweisen sowie alternative Optionen in Bezug auf Therapie oder Diagnose aufzuzeigen.Der Bericht wird in der Gebrauchsanweisung oder der Kennzeichnung ausgewiesen und wird der Öffentlichkeit über EUDAMED zugänglich gemacht. Dabei muss der Bericht so geschrieben sein, dass er auch für den Laien verständlich ist, sofern diese die Anwender sind.Vor Veröffentlichung erfolgt eine Validierung des Berichts durch die Benannte Stelle, die den Bericht dann in EUDAMED bereitstellt.Der SSCP wird jährlich aktualisiert.
Fazit
Im Überwachungszeitraum finden alle geplanten Aktivitäten zur Erfassung der Daten statt, anhand derer die fortgesetzte Leistungsfähigkeit und Sicherheit bewertet werden können. Gegen Ende dieses Zeitraums beginnt die zugehörige Berichterstattung. Diese Phase sollte kompakt gehalten werden, so dass sich keiner der Berichte im Übermaß verspätet. Während der Schreibphase bleibt das PMS-System selbstverständlich aktiv, und Pläne werden grundsätzlich geändert, sobald es nötig wird. Zusätzliche Aktivitäten werden den laufenden sofort hinzugefügt.Einen reibungslosen Ablauf, der die gegenseitige Bezugnahme der dabei erstellten Dokumente berücksichtigt, haben wir Ihnen hier vorgestellt. Im Umfang dieses Blogs können wir aber nicht jede individuelle Begebenheit berücksichtigen. Außerdem können jederzeit spezielle Situationen auftreten, wie
Wie können wir helfen?
Unsere Erfahrung zeigt, dass es zahlreiche Sondersituationen geben kann, die den angestrebten synchronisierten Rhythmus aller Berichte erschweren. Bei Ihnen ist dies der Fall? Unser Team erarbeitet mit Ihnen eine Strategie für Ihr Product Lifecycle Reporting, die Ihrem Produkt und Ihren Anforderungen entspricht und die ihnen hilft, auch auf Unvorhergesehenes souverän zu reagieren. Schreiben Sie uns eine Nachricht und finden Sie schnell und unkompliziert heraus, wie wir Ihnen weiterhelfen können!
Unsere Blogbeiträge werden mit höchster Sorgfalt recherchiert und erstellt, sind jedoch lediglich Momentaufnahmen in der Regulatorik, und diese ist in stetem Wandel. Wir gewährleisten nicht, dass ältere Inhalte noch aktuell und aussagekräftig sind. Wenn Sie nicht sicher sind, ob der Beitrag, den Sie auf dieser Seite gelesen haben, noch dem aktuellen Stand der Regulierung entspricht, nehmen Sie bitte Kontakt zu uns auf: Wir ordnen Ihr Thema schnell in den aktuellen Kontext ein.
Medical Device Expert
Regulatory Affairs & Technical Documentation




