
A Guide to Process Validation Part 2 - Smooth sailing, not stormy seas
21/05/2024
Do you have any questions about the article or would you like to find out more about our services? We look forward to hearing from you!Make a non-binding enquiry now
Ready for another journey into the world of process validation? In Part 1 of our Guide to Process Validation, we discussed the requirements of Annex I (General Safety and Performance Requirements (GSPR)) of Medical Device Regulation (EU) 2017/745 (MDR) and of ISO 13485:2016 for validation in production. We explored alternatives to validation and explained the ideal decision-making process. In this second part, we will apply the concepts introduced using an examplary process and provide detailed guidance on how to implement validation in practice.An important word in advance: For certain areas, specific normative requirements exist. If there are standards that regulate how validations (e.g., cleaning, disinfection, sterilization, packaging, or reprocessing) should be conducted for your product, these must, of course, be observed.
We will once again use the example from part 1 of our guide. It involves the company "Premier Components", their employee Vera Alid, and a filling and packaging process for which the customer, a manufacturer in the medical technology sector, has requested validation documentation. V. Alid reviews the work instructions for the products and speaks with the production manager to gain an overview of the processes carried out for the customer, Medical Devices LLC.The following description is the result of her research:
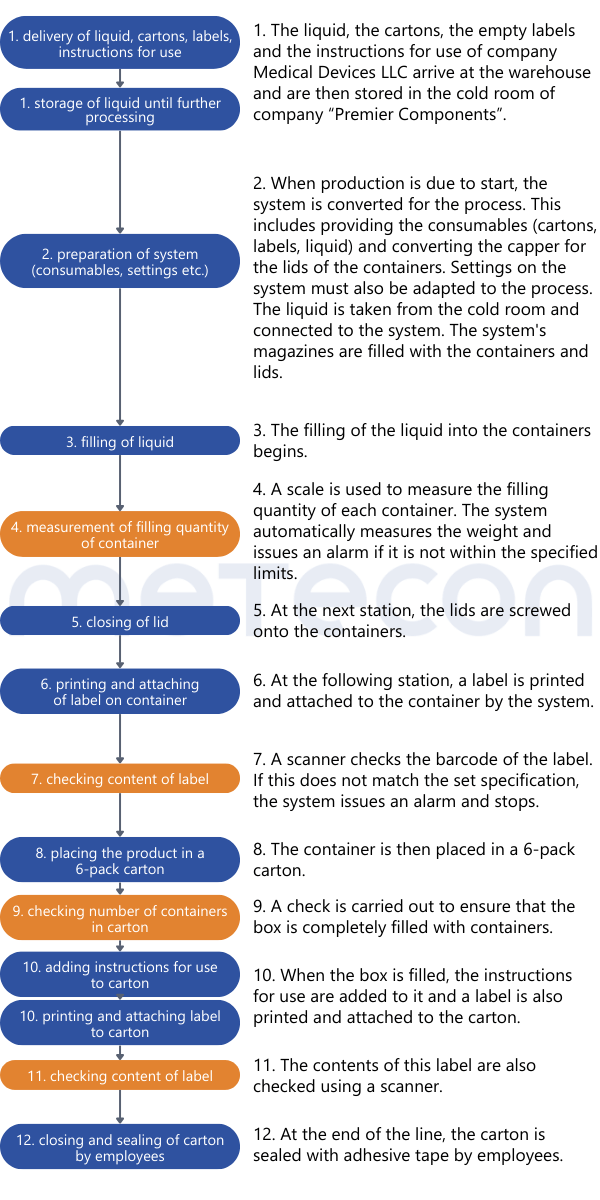
Once it is clear how the process is carried out, the analysis can begin. Since validation is potentially on the table, careful consideration must be given to which product attributes are relevant to safety and performance, how they are tested, and whether this testing can be classified as 100% verification. To aid this decision making process, it helps to list the process results or product attributes. Each attribute can be evaluated based on the process description to determine if and how it is verified. An example of such a table might look like this:
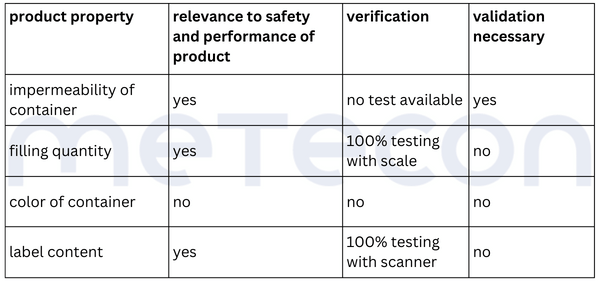
The liquid in the containers should not be able to escape and the containers should not be contaminated from the outside. This requirement dictates that the containers must be leak-proof. Since the impermeability of the container is not verified during the process and is critical to the product's safety and performance, it must be considered in the validation.From the weight measurement, it can be inferred that there is a requirement for the filling quantity of each container. This property is verified for each container by a scale, so that the property is 100% verified and does not need to be considered in a potential validation. However, as mentioned in part 1 of our guide, the reasons for this decision should be well documented.The color of the containers was determined to be irrelevant to the product's safety and performance. Whether and how it is verified is not relevant in the context of the validation decision.The content of the label was assessed as critical to the product's safety and performance. A scanner checks this information in each individual case. If there is any deviation from the specifications, the machine stops and triggers an alarm. Thus 100% verification is ensured and measures are in place to adequately reduce the risk of products with incorrect labels being processed further. However, proof must exist that this measure functions correctly (qualification).Practical Tip: As a supplier for medical device manufacturers, you should conduct the evaluation of the attribute's relevance for safety and performance together with the manufacturer. As a manufacturer, it is important that product experts involved in the decision-making process have sufficient product knowledge.
The process can be analyzed using risk analysis methods. These also provide evidence for the documentation of how the decision whether to test and how intensely to do this was made (statistics).To avoid confusion when referring to risk analysis in this article, please note: For process validation, risk analyses according to ISO 14971 are not required. You are free to choose the risk analysis method. What is important is that the probability of detection of errors can also be represented within the risk analysis. The risk analysis should also clearly identify which process parameters are present and which are critical. The term "parameter" does not solely refer to system settings but can be used for all process variables.After conducting a risk analysis, planning can begin on how to demonstrate that the identified risks have been mitigated. This brings us to the next step:
Operational qualification (OQ) is part of process validation and is defined in accordance with GHTF as "establishing by objective evidence that a process consistently produces a result or product meeting its predetermined requirements". This definition includes that it must be proven that the process still produces the required process results under challenge conditions or in the worst case, i.e. under the worst possible but still permitted conditions.Challenge conditions for a process must be determined individually in each case. Typical examples are: smallest or largest (packaging) variant, minimum or maximum process speed, variant with unfavorable materials, lowest or highest temperature.There can also be several challenge conditions for a parameter. A good example of this is the lower and upper temperature limits when sealing packaging: At the lower limit, the seal could be leaking or have insufficient strength. At the upper limit, the material could be damaged.ExampleThe filling quantity of the containers cannot be set within the system, only the "filling duration" parameter. (Assumption: There is also no 100 % check of the filling quantity within the system.) The filling quantity of the container has a specification with a lower and upper limit. The filling time could be set too short or too long so that the filling quantity specification is not met. If a range of 2-4 seconds is now permitted as a setting on the system, both the lower and upper limits of this setting must be considered during validation.In principle, all worst-case scenarios must be considered in the validation. For processes that are carried out with several different input variables (material, sizes, etc.), it is helpful to display the worst-case combinations in a matrix. This makes it easy to understand why certain worst-case combinations were selected. However, this matrix is not only helpful for the initial determination. Even if changes are made to the process or variables, this matrix can be used to determine the need for new OQ tests.Parameters for which only one setpoint exists during production (e.g., machine speed) are kept constant during the OQ. Settings in the OQ are not varied outside the prescribed process parameters. In such a case there is no specific "worst case."
OQ aims to demonstrate that the process produces acceptable results at the established limits. But how are these limits determined in the first place?Ideally, they should be established during process development. However, if such development reports are not available and the process has been running for a long time, how should one proceed?Employee V. Alid can imagine her boss's reaction if she suggests conducting development tests. In such a situation, using the current setpoint limits might be an option. The best step forward is to investigate whether worst-case combinations, such as the highest speed and lowest temperature in a sealing process, have been used in the past and if any issues arose. If there were indeed problems, they might reappear during the OQ, costing time and resources.In general, you should check if existing work instructions precisely specify the setpoints for critical parameters and if they align with the process parameter limits stated in the OQ. Knowledge of the process is crucial for planning the OQ and can save you from having to repeat tests.
Performance Qualification (PQ) is meant to prove that the procedure consistently produces a product meeting all predetermined requirements within nominal operating limites, i.e. under expected conditions (GHTF definition). These nominal operating limits are what needs to be determined for the process.Examples of variations in expected conditions can be found in both the direct input variables and the conditions during execution. Examples include: different material batches, variations in environmental conditions (temperature, humidity), variations in personnel conducting the process, setup changes in the equipment, general startup and shutdown of the equipment/process, and wear and tear of used tools.The possible variations and how they are to be evaluated in the context of the process to be carried out should be assessed in a risk analysis. In this way, you can also show that you have identified certain variations, but that further investigation of the effects is not necessary as part of a PQ. As is so often the case, you must have a sufficient basis of argumentation for this.With the OQ, the number of process runs to be carried out was determined by the number of worst-case scenarios. With PQ, this is somewhat more difficult to decide at first glance. Quite often, a production of three batches is chosen. For a long time, his was common practice in the area of pharmaceutical production . However, no such specification or recommendation can be found in the current regulations for medical devices (MDSAP AA, ISO 13485, GHTF).Here too, however, the magic words "risk-based approach" can help, because the actual aim is to "capture" the variation in conditions with the batches and then examine the results of these variations. In the end, the aim is to prove that the process result meets the required specifications. You can determine how many batches are required for these conditions as part of the risk analysis or PQ planning. If you are unsure, carrying out three batches is certainly a good guideline for orientation.Practical Tip: Suppose a process to be validated has been running for several years without changes. In such cases, you might check if enough data is available to conduct the PQ retrospectively. Data required would include evidence of process parameters or batches of input materials used during manufacturing. In a retrospective validation, you could list the possible variations of expected conditions and use the available data to show that these variations occurred in the past and produced products meeting the specifications. It is essential to evaluate any non-conformities in the process to show that even unfavorable data have been reviewed and assessed.The following graphic (based on a presentation by the U.S. Food and Drug Administration (FDA)) illustrates the perspectives of Process Development, Operational Qualification, and Performance Qualification. While the development phase broadly examines what is possible, sometimes pushing the technical limits of the equipment, the OQ and PQ phases increasingly align with the "normal" conditions during production.

Both Operational Qualification (OQ) and Performance Qualification (PQ) require the preparation of plans and reports. These reports should clearly show that the activities mentioned in the plan were carried out. Any deviations from the plan must be justified.For each validation project, higher-level documents can be created, serving as a control mechanism to document the necessary evidence in an organized manner. These documents can also describe the process and the results. Such a presentation allows for an easy introduction to the validation project during an audit, making the entire context clearer. This also means the subsequent OQ and PQ documents can be shorter. Additionally, a final "Validation Project Completion Report" can be created to list all the documents related to the project and to verify and highlight that all planned activities have been completed.For larger projects, these meta documents help to maintain an overview and later to convey this to auditors.Practical Tip: If you don't yet have a validation but need one, this is a good time to implement any planned changes to your process. Changes after the validation has been completed might require revalidation.When changes are made to the process, revalidation may become necessary. Changes could involve the material, the equipment used, the location where the process is conducted, or the process parameters. When planning a change, it must be evaluated whether this change impacts the validation status of the process. In any case, it is important to document the decision with justification and to involve experts in the evaluation. Sometimes, it may be sufficient to only partially revalidate the process. Perhaps only certain parts of the process are affected. If a thorough risk analysis of the process is available, this will help you immensely when evaluating such changes.What does this mean for our filling process? Let us assume that there is no 100% verification of the filling quantity. The specification for the container’s filling quantity is to be adjusted, increasing the upper limit. Since the quantity of fluid is influenced by the filling time parameter, this parameter's upper limit will also be increased. Tests in the OQ using the lower limit of the parameter do not need to be repeated, as this limit remains unchanged. Tests for seal integrity might also be omitted if it can be justified that a higher filling quantity has no impact on this parameter. Therefore, only tests with the upper parameter limit for the filling time appear necessary.It is also important to create a basis for the OQ when making process adjustments. Which process parameters are changed and to what extent when specifications or materials are changed is usually not clear. In such situations, tests are necessary to define process parameters and limits with sufficient certainty, which can then be used for OQ.
Our aim in this article was to highlight the importance of process and product knowledge for planning validation activities. Hopefully, you now have a clearer understanding of the activities behind the terms Operational Qualification (OQ) and Performance Qualification (PQ), and how you can convey your expertise in this area to auditors. We have not yet covered the qualification of devices, equipment, and systems, another critical aspect of process validation, which we will address in a future article.We know from frequent feedback of our newer clients that the topic of verification and validation can raise many questions, especially when encountered for the first time. We hope that our Guide to Process Validation offers you initial assistance in this area.If at any point in the process you are stuck, we will be happy to give you individual feedback on your specific questions.
 Leon Weißenhorn
Leon Weißenhorn
Step 1: Describing the process
We will once again use the example from part 1 of our guide. It involves the company "Premier Components", their employee Vera Alid, and a filling and packaging process for which the customer, a manufacturer in the medical technology sector, has requested validation documentation. V. Alid reviews the work instructions for the products and speaks with the production manager to gain an overview of the processes carried out for the customer, Medical Devices LLC.The following description is the result of her research:
Step 2: Analyzing the process
Once it is clear how the process is carried out, the analysis can begin. Since validation is potentially on the table, careful consideration must be given to which product attributes are relevant to safety and performance, how they are tested, and whether this testing can be classified as 100% verification. To aid this decision making process, it helps to list the process results or product attributes. Each attribute can be evaluated based on the process description to determine if and how it is verified. An example of such a table might look like this:
The liquid in the containers should not be able to escape and the containers should not be contaminated from the outside. This requirement dictates that the containers must be leak-proof. Since the impermeability of the container is not verified during the process and is critical to the product's safety and performance, it must be considered in the validation.From the weight measurement, it can be inferred that there is a requirement for the filling quantity of each container. This property is verified for each container by a scale, so that the property is 100% verified and does not need to be considered in a potential validation. However, as mentioned in part 1 of our guide, the reasons for this decision should be well documented.The color of the containers was determined to be irrelevant to the product's safety and performance. Whether and how it is verified is not relevant in the context of the validation decision.The content of the label was assessed as critical to the product's safety and performance. A scanner checks this information in each individual case. If there is any deviation from the specifications, the machine stops and triggers an alarm. Thus 100% verification is ensured and measures are in place to adequately reduce the risk of products with incorrect labels being processed further. However, proof must exist that this measure functions correctly (qualification).Practical Tip: As a supplier for medical device manufacturers, you should conduct the evaluation of the attribute's relevance for safety and performance together with the manufacturer. As a manufacturer, it is important that product experts involved in the decision-making process have sufficient product knowledge.
Risk analysis
The process can be analyzed using risk analysis methods. These also provide evidence for the documentation of how the decision whether to test and how intensely to do this was made (statistics).To avoid confusion when referring to risk analysis in this article, please note: For process validation, risk analyses according to ISO 14971 are not required. You are free to choose the risk analysis method. What is important is that the probability of detection of errors can also be represented within the risk analysis. The risk analysis should also clearly identify which process parameters are present and which are critical. The term "parameter" does not solely refer to system settings but can be used for all process variables.After conducting a risk analysis, planning can begin on how to demonstrate that the identified risks have been mitigated. This brings us to the next step:
Operational qualification (OQ)
Operational qualification (OQ) is part of process validation and is defined in accordance with GHTF as "establishing by objective evidence that a process consistently produces a result or product meeting its predetermined requirements". This definition includes that it must be proven that the process still produces the required process results under challenge conditions or in the worst case, i.e. under the worst possible but still permitted conditions.Challenge conditions for a process must be determined individually in each case. Typical examples are: smallest or largest (packaging) variant, minimum or maximum process speed, variant with unfavorable materials, lowest or highest temperature.There can also be several challenge conditions for a parameter. A good example of this is the lower and upper temperature limits when sealing packaging: At the lower limit, the seal could be leaking or have insufficient strength. At the upper limit, the material could be damaged.ExampleThe filling quantity of the containers cannot be set within the system, only the "filling duration" parameter. (Assumption: There is also no 100 % check of the filling quantity within the system.) The filling quantity of the container has a specification with a lower and upper limit. The filling time could be set too short or too long so that the filling quantity specification is not met. If a range of 2-4 seconds is now permitted as a setting on the system, both the lower and upper limits of this setting must be considered during validation.In principle, all worst-case scenarios must be considered in the validation. For processes that are carried out with several different input variables (material, sizes, etc.), it is helpful to display the worst-case combinations in a matrix. This makes it easy to understand why certain worst-case combinations were selected. However, this matrix is not only helpful for the initial determination. Even if changes are made to the process or variables, this matrix can be used to determine the need for new OQ tests.Parameters for which only one setpoint exists during production (e.g., machine speed) are kept constant during the OQ. Settings in the OQ are not varied outside the prescribed process parameters. In such a case there is no specific "worst case."
Excursus: Process development
OQ aims to demonstrate that the process produces acceptable results at the established limits. But how are these limits determined in the first place?Ideally, they should be established during process development. However, if such development reports are not available and the process has been running for a long time, how should one proceed?Employee V. Alid can imagine her boss's reaction if she suggests conducting development tests. In such a situation, using the current setpoint limits might be an option. The best step forward is to investigate whether worst-case combinations, such as the highest speed and lowest temperature in a sealing process, have been used in the past and if any issues arose. If there were indeed problems, they might reappear during the OQ, costing time and resources.In general, you should check if existing work instructions precisely specify the setpoints for critical parameters and if they align with the process parameter limits stated in the OQ. Knowledge of the process is crucial for planning the OQ and can save you from having to repeat tests.
Performance qualification (PQ)
Performance Qualification (PQ) is meant to prove that the procedure consistently produces a product meeting all predetermined requirements within nominal operating limites, i.e. under expected conditions (GHTF definition). These nominal operating limits are what needs to be determined for the process.Examples of variations in expected conditions can be found in both the direct input variables and the conditions during execution. Examples include: different material batches, variations in environmental conditions (temperature, humidity), variations in personnel conducting the process, setup changes in the equipment, general startup and shutdown of the equipment/process, and wear and tear of used tools.The possible variations and how they are to be evaluated in the context of the process to be carried out should be assessed in a risk analysis. In this way, you can also show that you have identified certain variations, but that further investigation of the effects is not necessary as part of a PQ. As is so often the case, you must have a sufficient basis of argumentation for this.With the OQ, the number of process runs to be carried out was determined by the number of worst-case scenarios. With PQ, this is somewhat more difficult to decide at first glance. Quite often, a production of three batches is chosen. For a long time, his was common practice in the area of pharmaceutical production . However, no such specification or recommendation can be found in the current regulations for medical devices (MDSAP AA, ISO 13485, GHTF).Here too, however, the magic words "risk-based approach" can help, because the actual aim is to "capture" the variation in conditions with the batches and then examine the results of these variations. In the end, the aim is to prove that the process result meets the required specifications. You can determine how many batches are required for these conditions as part of the risk analysis or PQ planning. If you are unsure, carrying out three batches is certainly a good guideline for orientation.Practical Tip: Suppose a process to be validated has been running for several years without changes. In such cases, you might check if enough data is available to conduct the PQ retrospectively. Data required would include evidence of process parameters or batches of input materials used during manufacturing. In a retrospective validation, you could list the possible variations of expected conditions and use the available data to show that these variations occurred in the past and produced products meeting the specifications. It is essential to evaluate any non-conformities in the process to show that even unfavorable data have been reviewed and assessed.The following graphic (based on a presentation by the U.S. Food and Drug Administration (FDA)) illustrates the perspectives of Process Development, Operational Qualification, and Performance Qualification. While the development phase broadly examines what is possible, sometimes pushing the technical limits of the equipment, the OQ and PQ phases increasingly align with the "normal" conditions during production.
General documentation of activities
Both Operational Qualification (OQ) and Performance Qualification (PQ) require the preparation of plans and reports. These reports should clearly show that the activities mentioned in the plan were carried out. Any deviations from the plan must be justified.For each validation project, higher-level documents can be created, serving as a control mechanism to document the necessary evidence in an organized manner. These documents can also describe the process and the results. Such a presentation allows for an easy introduction to the validation project during an audit, making the entire context clearer. This also means the subsequent OQ and PQ documents can be shorter. Additionally, a final "Validation Project Completion Report" can be created to list all the documents related to the project and to verify and highlight that all planned activities have been completed.For larger projects, these meta documents help to maintain an overview and later to convey this to auditors.Practical Tip: If you don't yet have a validation but need one, this is a good time to implement any planned changes to your process. Changes after the validation has been completed might require revalidation.When changes are made to the process, revalidation may become necessary. Changes could involve the material, the equipment used, the location where the process is conducted, or the process parameters. When planning a change, it must be evaluated whether this change impacts the validation status of the process. In any case, it is important to document the decision with justification and to involve experts in the evaluation. Sometimes, it may be sufficient to only partially revalidate the process. Perhaps only certain parts of the process are affected. If a thorough risk analysis of the process is available, this will help you immensely when evaluating such changes.What does this mean for our filling process? Let us assume that there is no 100% verification of the filling quantity. The specification for the container’s filling quantity is to be adjusted, increasing the upper limit. Since the quantity of fluid is influenced by the filling time parameter, this parameter's upper limit will also be increased. Tests in the OQ using the lower limit of the parameter do not need to be repeated, as this limit remains unchanged. Tests for seal integrity might also be omitted if it can be justified that a higher filling quantity has no impact on this parameter. Therefore, only tests with the upper parameter limit for the filling time appear necessary.It is also important to create a basis for the OQ when making process adjustments. Which process parameters are changed and to what extent when specifications or materials are changed is usually not clear. In such situations, tests are necessary to define process parameters and limits with sufficient certainty, which can then be used for OQ.
Conclusion
Our aim in this article was to highlight the importance of process and product knowledge for planning validation activities. Hopefully, you now have a clearer understanding of the activities behind the terms Operational Qualification (OQ) and Performance Qualification (PQ), and how you can convey your expertise in this area to auditors. We have not yet covered the qualification of devices, equipment, and systems, another critical aspect of process validation, which we will address in a future article.We know from frequent feedback of our newer clients that the topic of verification and validation can raise many questions, especially when encountered for the first time. We hope that our Guide to Process Validation offers you initial assistance in this area.If at any point in the process you are stuck, we will be happy to give you individual feedback on your specific questions.
Our blog posts are researched and created with the utmost care, but are only snapshots of the regulations, which are constantly changing. We do not guarantee that older content is still current or meaningful. If you are not sure whether the article you have read on this page still corresponds to the current state of regulation, please contact us: we will quickly place your topic in the current context.
Regulatory Affairs & Technical Documentation



