
Material Change, Sustainability, and Biological Safety of Medical Devices - A Trip into the Regulatory Jungle
31/05/2024
Do you have any questions about the article or would you like to find out more about our services? We look forward to hearing from you!Make a non-binding enquiry now
Since the Medical Device Regulation (EU) 2017/745 (MDR) came into effect in 2017, much has changed. For some companies, the MDR requirements continue to pose challenges, while others are already dealing with the implementation of numerous other guidelines and regulations that have arisen due to the MDR. In addition to these regulations, there are other considerations today, such as sustainability. In the current situation, particularly with regard to potential or necessary material changes to medical devices, it is necessary not only to adhere to current requirements, e.g. for biological safety, but to think several steps into the future to make such changes sustainable and economical. Let's venture together into the jungle of requirements.
The MDR’s main focus is on providing evidence of the safety and performance of a medical device throughout its entire lifecycle. The required evidence can be found in the general safety and performance requirements in Annex I of the MDR. Chapter II, Section 10.4 is entirely dedicated to the topic of "Substances" and addresses questions regarding which substances are permitted and which require special attention.In general, medical devices should be designed and manufactured in such a way that the risks from substances or particles that may be released from the device, including abrasion, degradation products, and processing residues, are reduced as far as possible.In the case of more critical medical devices which< 0.1% by mass . If such substances are to be used on a larger scale and if direct, invasive application is intended, a justification is required for their use, the contents of which are dealt with in sections 10.4.2 - 10.4.5 of the MDR.In principle, the latest scientific findings and other EU regulations or guidelines must always be taken into account in such a justification. These are then applied to the medical device in order to obtain a well-founded justification and assessment for the specific product. If critical substances are contained in higher mass percentages in the application parts of the medical device, the presence of such substances must be labeled on the products themselves or on the individual packaging. A precise list of the substances concerned is also required. Patient populations who are considered particularly susceptible to such substances and/or materials are informed about residual risks and any precautionary measures in the instructions for use.
If a change to a raw material is envisaged for a medical device, this does not necessarily mean that all previously valid tests to prove biocompatibility are no longer applicable. Rather, the assessment of such changes must be carried out beforehand as part of the risk management system in accordance with ISO 14971 (currently EN ISO 14971:2019+A11:2021). The potential effects of the change must be considered, which can provide different results depending on the intended change. If unacceptable risks are identified, manufacturers are obliged to specify these in a new version of the biological evaluation plan as part of the requirements in accordance with ISO 10993-1 (currently EN ISO 10993-1:2020). In addition, a scientific literature search must be carried out with regard to the intended new material and the intended clinical application of the medical device, as a well-founded statement on biological safety can be made only with sufficient physical and chemical characterization. Furthermore, the endpoints, which also result from EN ISO 10993-1 - Annex A, Table A.1, must be considered and evaluated in relation to the intended material change. If the risk is still in the unacceptable range or no suitable literature data can be found, then a new testing strategy is created with the subsequent final assessment as part of the creation of a new version of the biological assessment report.
In addition to the MDR and other requirements mentioned therein, the latest developments and decisions within the EU should always be considered, as these may have an impact on medical devices, even if the MDR is not directly affected. This can be seen, for example, in the current developments on sustainability as part of the EU Green Deal and the discussed restriction of perfluorinated and polyfluorinated alkyl substances (PFAS).The EU Green Deal aims to establish a modern, resource-efficient and competitive economy with the goal of zero net greenhouse gas emissions by 2050. Here, too, the medical device industry and the respective manufacturers must comply with regulations. The challenge here is to integrate what is technically feasible in a way that is viable from a regulatory and economic standpoint within the company.Part of the Green Deal is also the revision of Regulation (EC) No 1907/2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) and a new chemicals strategy in general. This aims to better protect people and the environment from harmful chemicals and to drive innovation by promoting the use of safer and more sustainable chemicals. To achieve this goal, a proposal to restrict an entire group of substances, PFAS, was submitted to the European Chemicals Agency (ECHA) in February 2023.PFAS have amphiphilic properties, i.e., they have hydrophobic carbon-fluorine compounds as well as hydrophilic parts due to the head group. Due to these amphiphilic properties in combination with their high heat resistance, the substances are chemically very inert and thus accumulate in the environment and can have far-reaching effects on human health.The production, placing on the market , and use of this group of substances is now to be successively restricted by the submitted proposal. The proposed ban on medical devices provides for a transitional regulation of 13.5 years (18 months plus 12-year exemption). Comments on the submitted draft could be submitted until September 2023 as part of a consultation process, which will be reviewed by ECHA's expert committees. The ECHA is expected to issue an opinion in 2024, although it is currently only possible to guess which changes will be introduced. Nevertheless, it is foreseeable that the entire PFAS substance group will be restricted and that there will only be exemptions for necessary areas, such as the medical technology industry. Therefore, the task of medical device manufacturers is now clear - to prepare for the upcoming ban in the best possible way.Regardless of whether it is about the future use of PFAS or more sustainable plastics such as bio-based or recycled plastics in medical devices, the targeted establishment of a meaningful data availability is always necessary.The following section outlines the various ways in which bio-based or recycled plastics can be used in medical devices in the future or how medical devices containing PFAS can be brought to market safely for the time being.
In order to strategically counter the impending restriction of PFAS, it is essential to list the product portfolio, including the materials used. If essential PFAS are identified, it is important to take direct action. The first step is always to communicate with all stakeholders at an early stage to ensure that suppliers, for example, continue to supply raw materials and do not discontinue all PFAS components due to the restriction. The origin of the full PFAS restriction lies not only in the critical properties but also in the frequent lack of data on the effects of PFAS on the environment and health, which is why it will be important to set up a well thought-out and customized assessment strategy for medical devices to adequately mitigate all potential risks. This strategy can be as follows:
 Figure 1: Possible evaluation strategy for medical devices; abbreviations: CMR = cancerogenic, mutagenic, toxic to reproduction, ED = endocrine disruptive
Figure 1: Possible evaluation strategy for medical devices; abbreviations: CMR = cancerogenic, mutagenic, toxic to reproduction, ED = endocrine disruptive
The strategy presented now offers the opportunity to carry out a data-based assessment of medical devices regarding their clinical use. In this way, proof can be provided that the use of medical devices does not pose any biological or toxicological risks despite the use of PFAS. In addition, further warnings and disposal instructions can be added to the instructions for use to limit uncontrolled release, which is currently the main problem, and thus enable a circular economy or at least controlled disposal of medical devices.This strategy can initially ensure that the safety and efficacy of medical devices containing PFAS that are already on the market is guaranteed. In the long term, however, it is crucial that the use of PFAS is reduced and that substitutes for PFAS are developed through research and development, which are considered for innovative medical devices as well as in devices already on the market.There are also other ways for manufacturers to prepare for an impending PFAS ban:
One objective of the EU Green Deal is to reduce net greenhouse gas emissions to achieve complete climate neutrality by 2050. However, the focus is also on decoupling growth from resource use in order to achieve the transition to a modern, resource-efficient and competitive economy while leaving no one, neither people nor regions, in the lurch. The EU Green Deal is closely linked to the term "sustainability". The dimensions of sustainability include environmental, social, and economic sustainability. These influence each other and must be given equal consideration.But how can the broad term "sustainability" be narrowed down for the medical device industry? One conceivable approach to complying with the contents of the Green Deal is the use of sustainable resources, such as
These two examples alone make it clear that the amount of evidence required will not decrease in the future. Even regarding the various regulations, there is still a battle to be fought through a jungle of requirements, which is presented somewhat more clearly below:
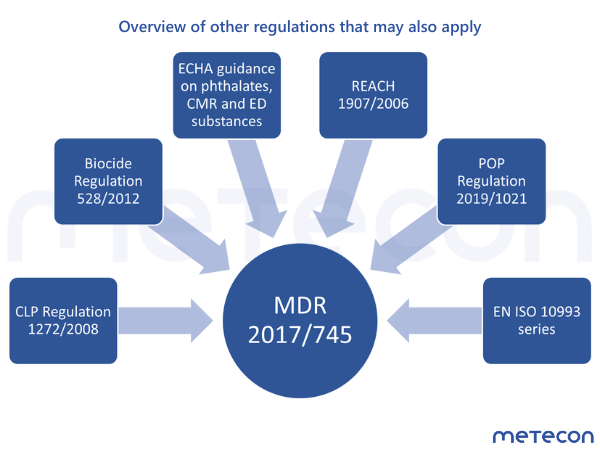 Figure 2: Overview of other regulations that may also apply. Abbreviations: MDR = Medical device regulation; CLP = Regulation on classification, labelling and packaging of substances and mixtures; ECHA = European Chemical Agency; REACH = Regulation on Registration, Evaluation, Authorization and Restriction of Chemicals; POP = Regulation on persistent organic pollutants; EN = European standard; ISO = International Organization for Standardization
Figure 2: Overview of other regulations that may also apply. Abbreviations: MDR = Medical device regulation; CLP = Regulation on classification, labelling and packaging of substances and mixtures; ECHA = European Chemical Agency; REACH = Regulation on Registration, Evaluation, Authorization and Restriction of Chemicals; POP = Regulation on persistent organic pollutants; EN = European standard; ISO = International Organization for Standardization
In the case of medical devices, the MDR remains the guiding regulation, which is why the proof of biological safety in accordance with the normative specifications of the EN ISO 10993 series does not change in principle. However, reference is made to other regulations or publications such as the ECHA. These requirements must be taken into account, and it must be ensured that suppliers comply with their duties under regulations like REACH. There are also constant innovations and amendments to additional regulations that may have an impact on medical devices, which is why the regulations must be addressed.
The future of medical devices and biological safety requirements will certainly remain exciting, especially in view of potential bans under REACH, which are likely to increase rather than decrease. Following the introduction of the MDR in 2017, companies initially failed to take action, which led to delays in the certification process. Lessons should now be derived from these mistakes, even if it initially appears to be a major, costly effort. Manufacturers should therefore now take action:
This article was commissioned by BIOPRO Baden-Württemberg GmbH. The state company BIOPRO Baden-Württemberg uses its expertise to support the transformation of the economy and society with a focus on the healthcare industry with its biotechnology, medical technology and pharmaceutical industries in Baden-Württemberg. As an innovation agency, it provides impetus for the development of the healthcare industry, initiates collaborations, accompanies innovations and start-ups and supports the sustainable transformation of healthcare-related supply and utilization chains. As the Regulatory Healthcare Industry BW contact point, funded by the Baden-Württemberg Ministry of Economic Affairs, Labor and Tourism, BIOPRO supports Baden-Württemberg companies in the healthcare industry in implementing new regulatory requirements. Further information on the activities of the Regulatory Healthcare Industry Contact Point: https://www.bio-pro.de/en/activities/anlaufstelle-regulatorik-gesundheitswirtschaft-bw
 Dr. Carolin Schilpp
Dr. Carolin Schilpp
Requirements of MDR for Materials and Substances
The MDR’s main focus is on providing evidence of the safety and performance of a medical device throughout its entire lifecycle. The required evidence can be found in the general safety and performance requirements in Annex I of the MDR. Chapter II, Section 10.4 is entirely dedicated to the topic of "Substances" and addresses questions regarding which substances are permitted and which require special attention.In general, medical devices should be designed and manufactured in such a way that the risks from substances or particles that may be released from the device, including abrasion, degradation products, and processing residues, are reduced as far as possible.In the case of more critical medical devices which
- are used invasively and have direct body contact
- (repeatedly) administer or remove medicinal products, body fluids, or other substances, including gases, from the body, or
- transport or store such medicinal products, body fluids, or other substances, including gases, which are (repeatedly) administered to the body,
- carcinogenic
- mutagenic
- toxic to reproduction
- or endocrine disruptive,
Influence of a (Raw) Material Change on Biological Safety
If a change to a raw material is envisaged for a medical device, this does not necessarily mean that all previously valid tests to prove biocompatibility are no longer applicable. Rather, the assessment of such changes must be carried out beforehand as part of the risk management system in accordance with ISO 14971 (currently EN ISO 14971:2019+A11:2021). The potential effects of the change must be considered, which can provide different results depending on the intended change. If unacceptable risks are identified, manufacturers are obliged to specify these in a new version of the biological evaluation plan as part of the requirements in accordance with ISO 10993-1 (currently EN ISO 10993-1:2020). In addition, a scientific literature search must be carried out with regard to the intended new material and the intended clinical application of the medical device, as a well-founded statement on biological safety can be made only with sufficient physical and chemical characterization. Furthermore, the endpoints, which also result from EN ISO 10993-1 - Annex A, Table A.1, must be considered and evaluated in relation to the intended material change. If the risk is still in the unacceptable range or no suitable literature data can be found, then a new testing strategy is created with the subsequent final assessment as part of the creation of a new version of the biological assessment report.
PFAS and sustainable synthetics – current options for use
In addition to the MDR and other requirements mentioned therein, the latest developments and decisions within the EU should always be considered, as these may have an impact on medical devices, even if the MDR is not directly affected. This can be seen, for example, in the current developments on sustainability as part of the EU Green Deal and the discussed restriction of perfluorinated and polyfluorinated alkyl substances (PFAS).The EU Green Deal aims to establish a modern, resource-efficient and competitive economy with the goal of zero net greenhouse gas emissions by 2050. Here, too, the medical device industry and the respective manufacturers must comply with regulations. The challenge here is to integrate what is technically feasible in a way that is viable from a regulatory and economic standpoint within the company.Part of the Green Deal is also the revision of Regulation (EC) No 1907/2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) and a new chemicals strategy in general. This aims to better protect people and the environment from harmful chemicals and to drive innovation by promoting the use of safer and more sustainable chemicals. To achieve this goal, a proposal to restrict an entire group of substances, PFAS, was submitted to the European Chemicals Agency (ECHA) in February 2023.PFAS have amphiphilic properties, i.e., they have hydrophobic carbon-fluorine compounds as well as hydrophilic parts due to the head group. Due to these amphiphilic properties in combination with their high heat resistance, the substances are chemically very inert and thus accumulate in the environment and can have far-reaching effects on human health.The production, placing on the market , and use of this group of substances is now to be successively restricted by the submitted proposal. The proposed ban on medical devices provides for a transitional regulation of 13.5 years (18 months plus 12-year exemption). Comments on the submitted draft could be submitted until September 2023 as part of a consultation process, which will be reviewed by ECHA's expert committees. The ECHA is expected to issue an opinion in 2024, although it is currently only possible to guess which changes will be introduced. Nevertheless, it is foreseeable that the entire PFAS substance group will be restricted and that there will only be exemptions for necessary areas, such as the medical technology industry. Therefore, the task of medical device manufacturers is now clear - to prepare for the upcoming ban in the best possible way.Regardless of whether it is about the future use of PFAS or more sustainable plastics such as bio-based or recycled plastics in medical devices, the targeted establishment of a meaningful data availability is always necessary.The following section outlines the various ways in which bio-based or recycled plastics can be used in medical devices in the future or how medical devices containing PFAS can be brought to market safely for the time being.
Per- and polyfluorinated alkyl substances – PFAS
In order to strategically counter the impending restriction of PFAS, it is essential to list the product portfolio, including the materials used. If essential PFAS are identified, it is important to take direct action. The first step is always to communicate with all stakeholders at an early stage to ensure that suppliers, for example, continue to supply raw materials and do not discontinue all PFAS components due to the restriction. The origin of the full PFAS restriction lies not only in the critical properties but also in the frequent lack of data on the effects of PFAS on the environment and health, which is why it will be important to set up a well thought-out and customized assessment strategy for medical devices to adequately mitigate all potential risks. This strategy can be as follows:
Figure 1: Possible evaluation strategy for medical devices; abbreviations: CMR = cancerogenic, mutagenic, toxic to reproduction, ED = endocrine disruptiveThe strategy presented now offers the opportunity to carry out a data-based assessment of medical devices regarding their clinical use. In this way, proof can be provided that the use of medical devices does not pose any biological or toxicological risks despite the use of PFAS. In addition, further warnings and disposal instructions can be added to the instructions for use to limit uncontrolled release, which is currently the main problem, and thus enable a circular economy or at least controlled disposal of medical devices.This strategy can initially ensure that the safety and efficacy of medical devices containing PFAS that are already on the market is guaranteed. In the long term, however, it is crucial that the use of PFAS is reduced and that substitutes for PFAS are developed through research and development, which are considered for innovative medical devices as well as in devices already on the market.There are also other ways for manufacturers to prepare for an impending PFAS ban:
- modulation of PFAS, e.g. to enable faster, toxicologically safe degradation
- modulation of medical devices to limit the potential release of PFAS
- manufacturers and suppliers can apply for a medical exemption for the necessary substances (time-consuming)
- registration and authorization of the substances with the competent authority as an interest group of several manufacturers and suppliers/subcontractors
More sustainable synthetics in medical products
One objective of the EU Green Deal is to reduce net greenhouse gas emissions to achieve complete climate neutrality by 2050. However, the focus is also on decoupling growth from resource use in order to achieve the transition to a modern, resource-efficient and competitive economy while leaving no one, neither people nor regions, in the lurch. The EU Green Deal is closely linked to the term "sustainability". The dimensions of sustainability include environmental, social, and economic sustainability. These influence each other and must be given equal consideration.But how can the broad term "sustainability" be narrowed down for the medical device industry? One conceivable approach to complying with the contents of the Green Deal is the use of sustainable resources, such as
- Recycled synthetics: These consist of reprocessed synthetics waste, which is usually obtained in a chemical or mechanical recycling process. With recycled synthetics, a distinction is made between post-consumer recyclates – recycled synthetics from household waste – and post-industrial recyclates – recycled synthetics from "waste" from other industries such as edible oil refineries,
- Bio-based synthetics: These are synthetics made from renewable raw materials,
- Biodegradable synthetics: Biodegradable synthetics include all synthetics that can be decomposed by microorganisms.
- The use of purely bio-based synthetics is not yet commonplace, but initial approaches are already finding their way into practice. Nevertheless, mixtures of fossil and bio-based synthetics are currently mostly used in varying proportions. The reason for the mixture is that this is usually the only way to achieve the desired properties, such as stability, purity, and thermal and mechanical resilience. However, there are also polymers such as polyethylene, polylactic acids (polylactides), polyhydroxyalkanoates (PAH), polybutylene succinate (PBS) or poly (hydroxybutyrate-co-hydroxyvalerate) copolymers (PHBV), which can be used 100 % bio-based.
- While the use of synthetics with a variable proportion of recycled synthetics is already possible on a case-by-case basis for medical device packaging, for example, the use of recyclates for higher-classified medical products is currently almost impossible without standardization in the production of recyclates, e.g. through consistent quality and always the same proportion of recyclates.
Requirements for Verification – What is Really Necessary?
These two examples alone make it clear that the amount of evidence required will not decrease in the future. Even regarding the various regulations, there is still a battle to be fought through a jungle of requirements, which is presented somewhat more clearly below:
Figure 2: Overview of other regulations that may also apply. Abbreviations: MDR = Medical device regulation; CLP = Regulation on classification, labelling and packaging of substances and mixtures; ECHA = European Chemical Agency; REACH = Regulation on Registration, Evaluation, Authorization and Restriction of Chemicals; POP = Regulation on persistent organic pollutants; EN = European standard; ISO = International Organization for StandardizationIn the case of medical devices, the MDR remains the guiding regulation, which is why the proof of biological safety in accordance with the normative specifications of the EN ISO 10993 series does not change in principle. However, reference is made to other regulations or publications such as the ECHA. These requirements must be taken into account, and it must be ensured that suppliers comply with their duties under regulations like REACH. There are also constant innovations and amendments to additional regulations that may have an impact on medical devices, which is why the regulations must be addressed.
Conclusion
The future of medical devices and biological safety requirements will certainly remain exciting, especially in view of potential bans under REACH, which are likely to increase rather than decrease. Following the introduction of the MDR in 2017, companies initially failed to take action, which led to delays in the certification process. Lessons should now be derived from these mistakes, even if it initially appears to be a major, costly effort. Manufacturers should therefore now take action:
- Get an overview of raw materials
- List all potentially affected raw materials/medical devices that would be affected by changes due to bans under REACH, such as the PFAS ban, or where the use of sustainable materials is being sought
- Prioritization according to the importance or necessity of the medical devices: For example, if treatable diseases can no longer be treated due to the ban on PFAS, then such products have priority.
- Prioritization according to the need to use the prohibited substances/materials and the need for change
- Create a basis for argumentation through comprehensive risk analysis and characterization of the intended substances and medical devices
- Find "allies" for interest groups and applying for medical exemptions
- Sustainability can take place on many levels and does not always have to start directly at the most critical point, such as a complete change of material: Reducing the amount of fossil raw materials in the product itself would be a conceivable approach.
- Use of bio-based or recycled synthetics for use in medical devices for components without direct physical contact as a first step, as otherwise it is difficult to achieve a sensible cost-benefit ratio. This point should also be considered from the outset when developing new medical devices.
This article was commissioned by BIOPRO Baden-Württemberg GmbH. The state company BIOPRO Baden-Württemberg uses its expertise to support the transformation of the economy and society with a focus on the healthcare industry with its biotechnology, medical technology and pharmaceutical industries in Baden-Württemberg. As an innovation agency, it provides impetus for the development of the healthcare industry, initiates collaborations, accompanies innovations and start-ups and supports the sustainable transformation of healthcare-related supply and utilization chains. As the Regulatory Healthcare Industry BW contact point, funded by the Baden-Württemberg Ministry of Economic Affairs, Labor and Tourism, BIOPRO supports Baden-Württemberg companies in the healthcare industry in implementing new regulatory requirements. Further information on the activities of the Regulatory Healthcare Industry Contact Point: https://www.bio-pro.de/en/activities/anlaufstelle-regulatorik-gesundheitswirtschaft-bw
Our blog posts are researched and created with the utmost care, but are only snapshots of the regulations, which are constantly changing. We do not guarantee that older content is still current or meaningful. If you are not sure whether the article you have read on this page still corresponds to the current state of regulation, please contact us: we will quickly place your topic in the current context.
Biological Safety Expert
Regulatory Affairs & Technical Documentation



