
UPDATE: Brexit, UKCA und MHRA: Wichtige Änderungen und deren Fristen
15.01.2021
Sie haben Fragen zum Beitrag oder möchten mehr über unsere Leistungen erfahren? Wir freuen uns auf Ihre Nachricht!Jetzt unverbindlich anfragen
(Beachten Sie bitte auch unseren Beitrag vom 26.07.2021: Post-Brexit: Die neu gültige Gesetzgebung und bisherige Erfahrungen)Am 1. Januar 2021 endete die Übergangsfrist für den Brexit. Bruntje Esrom, Quality Management Expert bei Metecon, gibt Antworten auf folgende Fragen: Welche Schritte für einen Marktzugang in GB sind jetzt erforderlich? Und welche weiteren Hürden müssen die Hersteller von Medizinprodukten und IVD nehmen? Die britische Regulierungsbehörde für Arzneimittel und Gesundheitsprodukte (MHRA) hat zum 01.01.2021 insgesamt 48 neue Leitfäden zu den Prozessen veröffentlicht, die nach Ablauf der Brexit-Übergangsfrist Anwendung finden. Vor allem für Medizinprodukte müssen die Fristen der regulatorischen Neuerungen im Auge behalten werden, um auch nach Ablauf der Übergangsfrist den reibungslosen Marktzugang in GB aufrechtzuerhalten. Generell dürfen CE-gekennzeichnete Medizinprodukte nach Richtlinie 92/42/EWG (MDD) und IVD bis 30.06.2023 in GB in Verkehr gebracht werden; bis zu diesem Stichtag findet die CE-Kennzeichnung in GB Anerkennung. Hersteller von Medizinprodukten und IVD sollten aber rechtzeitig agieren, um die neuen Zulassungs- und Kennzeichnungsanforderungen fristgerecht implementieren zu können. Eine Neuerung der Kennzeichnungsanforderungen ist, dass das bisher einheitliche CE-Kennzeichen, das bislang in nahezu allen europäischen Zielmärkten Anerkennung fand, durch eine eigene Kennzeichnung ersetzt werden soll. Dieses UKCA-Kennzeichen wurde eigens für den Marktzugang in GB entwickelt und wird künftig gefordert.
Allerdings ersetzt andersherum die UKCA-Kennzeichnung die Kennzeichnung des Produkts mit dem CE-Kennzeichen für andere europäische Zielmärkte NICHT. Demnach müssen hier beide Verfahren angewandt werden, um den regulatorischen Anforderungen der gewünschten Zielmärkte (UK und EU) gerecht zu werden. Bereits im Registrierungsprozess und CE-Konformitätsbewertungsverfahren befindliche Produkte, deren Verfahren noch vor dem 01.01.2021 bei der MHRA gestartet wurden, werden automatisch im Konformitätsbewertungsverfahren für die UKCA Kennzeichnung weitergeführt. Dies stellt sicher, dass Hersteller den Zulassungsprozess für den Markzugang in GB nicht erneut starten müssen und damit keine Zeit verlieren.
Für größere Ansicht Grafik anklicken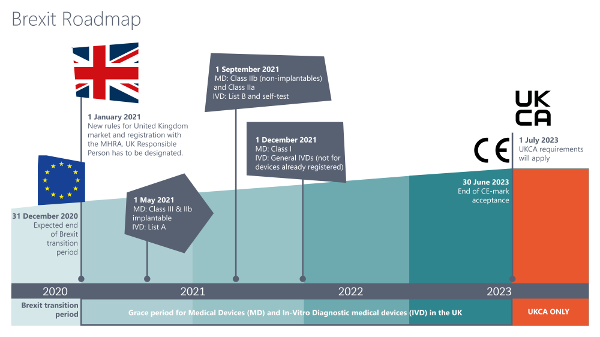 © 2020/12 Metecon GmbH
© 2020/12 Metecon GmbH
Quelle: Regulating medical devices from 1 January 2021 - GOV.UK (www.gov.uk)
Bestimmungen und Fristen für die UKCA-Kennzeichnung ab 01.01.2021
Die Roadmap für den Marktzugang im UK sieht vor, dass alle Medizinprodukte und IVD ab dem 01.01.2021 nach den neuen Registrierungs- und Kennzeichnungsrichtlinien in Verkehr gebracht werden müssen – je nach Klassifizierung gelten hierfür allerdings Kulanzfristen für die Umsetzung.- Für bereits im UK-Markt befindliche Klasse III- und IIb-Implantate, sowie für aktive, implantierbare Medizinprodukte und IVD gemäß Liste A gilt eine Umstellungspflicht der Kennzeichnung ab 01.05.2021,
- Für andere Klasse IIb- und IIa-Produkte und IVD gemäß Liste B und IVD zur Eigenanwendung gilt die neue Kennzeichnungspflicht ab 01.09.2021,
- Klasse I-Produkte und sonstige IVD müssen ab dem 01.01.2022 nach den neuen geltenden Richtlinien gekennzeichnet sein und registriert werden.
Allerdings ersetzt andersherum die UKCA-Kennzeichnung die Kennzeichnung des Produkts mit dem CE-Kennzeichen für andere europäische Zielmärkte NICHT. Demnach müssen hier beide Verfahren angewandt werden, um den regulatorischen Anforderungen der gewünschten Zielmärkte (UK und EU) gerecht zu werden. Bereits im Registrierungsprozess und CE-Konformitätsbewertungsverfahren befindliche Produkte, deren Verfahren noch vor dem 01.01.2021 bei der MHRA gestartet wurden, werden automatisch im Konformitätsbewertungsverfahren für die UKCA Kennzeichnung weitergeführt. Dies stellt sicher, dass Hersteller den Zulassungsprozess für den Markzugang in GB nicht erneut starten müssen und damit keine Zeit verlieren.
Für größere Ansicht Grafik anklicken
© 2020/12 Metecon GmbHQuelle: Regulating medical devices from 1 January 2021 - GOV.UK (www.gov.uk)
Beantragung der Registrierung der neu gekennzeichneten Produkte bei der MHRA
Die Kennzeichnungs- und Registrierungspflicht seit dem 01.01.2021 beinhaltet, dass die Hersteller die Produkte, die sie auf dem UK-Markt in Verkehr bringen möchten, bei der MHRA registrieren müssen. Die EU wird nach der Übergangsfrist die Benannten Stellen im UK anerkennen, allerdings wird es den Benannten Stellen innerhalb des UK nicht mehr möglich sein, CE-Zertifikate nach MDR auszustellen. Eine Ausnahme gilt für die CE-UKNI-Kennzeichnung, die ihre Gültigkeit in Nordirland behält. Die Vergabe der UKCA-Kennzeichnung erfolgt künftig durch designierte UK-Benannte Stellen. Seit dem 01.01.2021 ist es der MHRA möglich, diese designierten UK-Benannten Stellen zu benennen. Eine Ausnahme gilt für Klasse I-Produkte, die durch Hersteller selbst zertifiziert werden können. Klasse Is-Produkte benötigen aber ebenfalls die Zustimmung durch die UK-Benannte Stelle.Wer ist befugt, Medizinprodukte und IVD bei der MHRA zu registrieren?
Bei der Registrierung der Medizinprodukte und IVD kommt eine weitere Neuerung ins Spiel, mit der sich Hersteller künftig auseinandersetzen müssen. Hersteller außerhalb des UK, die Zugang zum britischen Markt erlangen wollen, müssen eine sogenannte „UK Responsible Person“ benennen und installieren, die seit dem 01.01.2021 durch die neue Rechtslage nach dem Brexit gefordert ist. Diese UK Responsible Person übernimmt sowohl die Verantwortung für die im UK in Verkehr gebrachten Produkte als auch für die Registrierung der Medizinprodukte und IVD bei der MHRA.Stichtage der Registrierungspflicht bei der MHRA nach Produkten:- 01.05.2021 für Hersteller von Klasse III- und IIb-Medizinprodukten, Liste A-IVD-Produkte und aktiven, implantierbaren Medizinprodukten,
- 01.09.2021 für Hersteller von anderen Klasse IIb- und IIa-Produkten und Liste B-IVD-Produkten sowie Selbsttest-IVD,
- 01.01.2022 für Hersteller von Klasse I-Produkten.
Die künftigen regulatorischen Regelwerke für UK
Die EU-MDR und EU-IVDR gelten zwar zunächst während der Übergangsfrist bis zum 30.06.2023, jedoch strebt das UK die Erstellung eines eigenen Regelwerks, des UK MMD Bill, an. Dieser Gesetzesentwurf wird aktuell im House of Lords des britischen Parlaments geprüft. Experten erwarten eine Verabschiedung des Gesetzes jedoch nicht vor Anfang 2022. Eine weitere Hürde stellen die regulatorischen Anforderungen des künftig für den britischen Markt benötigten Konformitätsbewertungsverfahrens dar. Die Anforderung für die Vergabe der UKCA-Kennzeichnung soll sich demnach auf die relevanten Anhänge der EU-AIMDD, -MDD und -IVDD stützen. Allerdings befinden sich die Hersteller gerade mitten in der Umstellung auf die EU-MDR und -IVDR und müssen sich nun trotzdem weiterhin an den Vorgaben der EU-AIMDD, -MDD und -IVDD orientieren. So müssen neben den neuen EU-Vorgaben auch die künftigen Vorgaben für GB erfüllt werden.Resümee
Auf Sie als Hersteller von Medizinprodukten oder IVD kommen mit dem Brexit einige wichtige und aufwändige Neuerungen zu. Unabdingbar ist deshalb der Blick auf die zu beachtenden Fristen und die neuen regulatorischen Anforderungen durch MHRA und die britische Regierung. Noch weiß keiner, wie die regulatorischen Regelwerke, die die EU-MDD und EU-IVD letztendlich ablösen sollen, tatsächlich aussehen und wann diese zur Verfügung stehen werden. Auch die Frage danach, wie schnell die Installation der UK-Benannten Stellen erfolgt und wie reibungslos die Registrierung von Produkten möglich sein wird, können wir heute leider noch nicht beantworten. Der Brexit ist vollzogen und vieles ist ungeklärt. Umso wichtiger ist es für Hersteller, so gut als möglich vorbereitet zu sein auf das, was da auf sie zukommt. Heute bereits Strategien zu entwickeln, wie auf verschiedene Szenarien reagiert und im Sinne einer künftigen Positionierung am britischen Markt agiert werden kann und will, scheint unerlässlich.Was Sie als Hersteller zum jetzigen Zeitpunkt tun sollten, ist eine UK Responsible Person zu benennen und diese bei der MHRA zu registrieren, um weiterhin Ihre bereits im Markt befindlichen Medizinprodukte in GB vertreiben zu können und die Einführung von neuen Medizinprodukten nicht zu verzögern.Sie haben Fragen zum Thema? Rufen Sie mich an oder schreiben Sie mir. Ich freue mich darauf, Sie kennenzulernen!Herzliche Grüße Quellen:- https://www.gov.uk/government/collections/new-guidance-and-information-for-industry-from-the-mhra
- https://nibsc.org/about_us/latest_news/guidance_for_mfrs.aspx
Unsere Blogbeiträge werden mit höchster Sorgfalt recherchiert und erstellt, sind jedoch lediglich Momentaufnahmen in der Regulatorik, und diese ist in stetem Wandel. Wir gewährleisten nicht, dass ältere Inhalte noch aktuell und aussagekräftig sind. Wenn Sie nicht sicher sind, ob der Beitrag, den Sie auf dieser Seite gelesen haben, noch dem aktuellen Stand der Regulierung entspricht, nehmen Sie bitte Kontakt zu uns auf: Wir ordnen Ihr Thema schnell in den aktuellen Kontext ein.




